
Abstract
Matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) is a powerful tool for investigating the distribution of proteins and small molecules within biological systems through the in situ analysis of tissue sections. MALDI-IMS can determine the distribution of hundreds of unknown compounds in a single measurement and enables the acquisition of cellular expression profiles while maintaining the cellular and molecular integrity. In recent years, a great many advances in the practice of imaging mass spectrometry have taken place, making the technique more sensitive, robust, and ultimately useful. In this review, we focus on the current state of the art of MALDI-IMS, describe basic technological developments for MALDI-IMS of animal and human tissues, and discuss some recent applications in basic research and in clinical settings.
Similar content being viewed by others


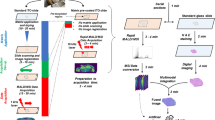
Introduction
In biomedical research, the discovery of new biomarkers and new drugs demands analytical techniques with high sensitivity together with increased throughput. The possibility to localize or to follow changes in organisms at the molecular level by imaging component distributions of specific tissues is of prime importance to unravel biochemical pathways and develop new treatments and drugs (Rohner et al. 2005). In recent years, several proteomic methodologies have been developed that now make it possible to identify, characterize, and comparatively quantify the relative level of expression of hundreds of proteins that are co-expressed in a given cell type or tissue. These advances have resulted from the integration of diverse scientific disciplines including molecular and cellular biology, protein/peptide chemistry, bioinformatics, analytical and bioanalytical chemistry, and the use of instrumental and software tools. Mass spectrometry has become an indispensable tool for proteomic studies (Lavoie and Paiement 2008; Chaurand et al. 2004a; Aebersold and Goodlett 2001; Godovac-Zimmermann and Brown 2001; Lahm and Langen 2000; McDonald and Yates 2000; Pandey and Mann 2000; Roepstorff 1997). Desorption and ionization techniques such as matrix-assisted laser desorption ionization mass spectrometry (MALDI MS) (Hillenkamp et al. 1991; Karas and Hillenkamp 1988) and electrospray ionization mass spectrometry (ESI MS) (Fenn et al. 1989) have revolutionized the analysis of proteins. These improvements offer levels of mass accuracy and sensitivity never before achieved for the detection and identification of proteins. MALDI MS is an ideal tool to investigate complex protein mixtures. It utilizes a matrix, a small acidic aromatic molecule that absorbs energy at the wavelength of the irradiating laser. The analyte molecule is mixed with the matrix, deposited on a target plate or a conductive glass slide and allowed to dry. During the drying process, matrix-analyte co-crystals form. These crystals are then submitted to very short laser pulses (typically UV laser light), resulting in the desorption and ionization of the analyte molecule. The ions mass-to-charge (m/z) are measured in a time-of-flight (TOF) mass analyzer (Chaurand et al. 2005).
To date, there are several technologies available to analyze proteins in a tissue specimen. The most commonly used technology is the separation and visualization of proteins by 2-dimensional (2D) gel electrophoresis and subsequent identification by mass spectrometry and database searching (Lahm and Langen 2000; Godovac-Zimmermann and Brown 2001). One of the drawbacks of the 2-D gel technology is that sample preparation removes the direct relationship between morphological tissue regions and a specific protein. One solution to this problem is to purify cells from thin tissue sections by microdissection techniques prior to protein extraction. Although such an approach has been successful (Curran et al. 2000), the extraction of a sufficient quantity of material is very labor intensive and requires a large amount of microdissected cells. A major advantage of direct MALDI analysis is to avoid time-consuming extraction, purification or separation steps, which have the potential for producing artefacts. One of the recent applications of MALDI MS is its use to profile and image proteins directly from tissue sections (Caprioli et al. 1997; Stoeckli et al. 2001; Todd et al. 2001; Chaurand and Caprioli 2002; Chaurand et al. 2002, 2004b, 2005; Cornett et al. 2007).
MALDI imaging mass spectrometry (MALDI-IMS) is a new technology that allows for simultaneous mapping of hundreds of peptides and proteins present in thin tissue sections with a lateral resolution of approximately 30–50 μm. Matrix is first uniformly deposited over the surface of the section, utilizing procedures optimized to minimize protein migration. Proteins are then desorbed from discrete spots or pixels upon irradiation of the sample in an ordered array or raster of the surface. Each pixel thus is keyed to a full mass spectrum consisting of signals from protonated species of molecules desorbed from that tissue region. A plot of the intensity of any one signal produces a map of the relative amount of that compound over the entire imaged surface. This technology provides a powerful discovery tool for the investigation of biological processes because the identities of proteins observed do not need to be known in advance. MALDI-IMS under various forms has already been successfully used to characterize the expression of proteins and other organic biological compounds in numerous normal and diseased tissues (Chaurand et al. 2005). For example, protein organization in mouse colon (Chaurand et al. 1999), brain (Stoeckli et al. 2001; Todd et al. 2001; Chaurand et al. 2002, 2004b) and epididymis (Chaurand et al. 2003) as well as phospholipid organization in mammalian lens tissue (Rujoi et al. 2004) has been studied. Variations in protein expression have been investigated in the cases of Parkinson’s (Pierson et al. 2004) and Alzheimer’s (Stoeckli et al. 2002) diseases. Several forms of cancers have also been investigated including gliomas (Stoeckli et al. 2001; Chaurand et al. 2004b; Schwartz et al. 2004), breast cancer (Palmer-Toy et al. 2000; Xu et al. 2002), prostate cancer (Masumori et al. 2001, Schwamborn et al. 2007), colon cancer (Chaurand et al. 2001) and lung cancer (Bhattacharya et al. 2003; Yanagisawa et al. 2003). In this later study (Yanagisawa et al. 2003), protein patterns have been shown to be predictive of diagnosis and prognosis. Methodologies aimed at detecting and mapping pharmaceutical compounds by direct MALDI MS analysis of sections from dosed tissues have also been described (Hsieh et al. 2007; Khatib-Shahidi et al. 2006; Reyzer et al. 2003).
This review article provides an overview of several animal and human tissue imaging applications performed by MALDI-IMS, including sample preparation, matrix selection and application, histological staining in MALDI analysis, tissue profiling, imaging, and data analysis. Several applications represent direct translation of this technology to basic research and in clinical applications.
In situ analysis of proteins from tissue sections
Instrumentation
To use a mass spectrometer as an imaging instrument, it is essential for the spectrometer to be equipped with an automatic rastering function, automatic data acquisition system, and visualization software. Recently, several manufacturers have released novel instruments having these features. Almost all manufacturers have developed in-house software for their instruments and have included a driver for instrument and image reconstruction. Instruments used for imaging mass spectrometry can be classified according to how ions are generated from the sample: either by irradiation by pulsed laser or bombardment by energetic particles. Laser based systems include MALDI and laser desorption ionization instruments. Secondary ion mass spectrometry (SIMS) systems use particle bombardment with a continuous beam of highly-focused, energetic ions. In general, these imaging instruments were based mostly on MALDI-TOF, MALDI-TOF/TOF, or TOF-SIMS. MALDI-TOF imaging instruments are high throughput, and TOF-SIMS imaging instruments can provide submicrometer spatial resolution of ~500 nm (Altelaar et al. 2006). Both instruments have powerful advantages in imaging mass spectrometry. There are several published articles discussing and comparing MALDI and SIMS imaging (Heeren et al. 2005; McDonnell and Heeren 2007; Shimma et al. 2008). This review focuses on MALDI imaging.
Tissue sample acquisition and preparation
The representativity of an imaging mass spectrometry experiment, in terms of both chemical representativity and spatial integrity, can only be as good as the worst step in the entire process of sample acquisition, preparation, imaging mass spectrometry, and interpretation. In all sample-handling steps throughout acquisition and preparation, the spatial and chemical integrity must be maintained, within a length scale determined by the spatial resolution of the mass spectrometry technique used (McDonnell et al. 2007). Preparation methods for MALDI-IMS tissue imaging must be carefully performed to maintain the spatial arrangement of compounds and avoid delocalization and degradation of the analytes. Experimental parameters that should be considered include treatment of tissue immediately after sample procurement, sectioning, sample transfer to the MALDI target plate or conductive glass slide, matrix application, and tissue storage after sectioning (Caldwell and Caprioli 2005). Careful handling of tissue samples, including freezing the tissue in liquid nitrogen immediately after procurement, is essential to preserve the native condition of the tissue. Typically, the interior of tissue samples is exposed using cryosectioning techniques. Embedding in optimal cutting temperature (OCT) polymer or Tissue-Tek® should be avoided as it can smear across the sample, thus compromising the chemical integrity. Embedding in gelatine (Altelaar et al. 2005) and agarose (Kruse and Sweedler 2003) has been used to facilitate handling of small or fragile samples (e.g., biopsies) but most often cryosectioning is performed directly with the frozen tissue samples (Kruse and Sweedler 2003; Schwartz et al. 2003). Although thickness is not critical, 10–20 μm thick sections are optimal for handling and analyzing in the high vacuum environment of the mass spectrometer. The thickness of the tissue sections used is normally 10–20 μm: thin sections can be very fragile and difficult to manipulate, whereas thicker sections need longer to dry and, because they are insulating samples, can adversely affect the performance of the mass analyzer (Caldwell and Caprioli 2005). The sample plate of choice is a transparent conductive slide such as indium-tin-oxide-coated glass slides because they allow the optical microscopic image acquisition and mass spectrometry to be performed on the same sample (Chaurand et al. 2004b; Altelaar et al. 2005).
The significant potential of tissue-based proteomic biomarker studies can be restricted by difficulties in accessing tissue samples in optimal fresh-frozen form. While archival formalin-fixed tissue collections with attached clinical and outcome data represent a valuable alternate resource, the use of formalin as a fixative which induces protein cross-linking, has generally been assumed to render them unsuitable for proteomic studies. Formalin fixation of tissues, generally followed by paraffin embedding, is the standard and well-established processing method employed by pathologists. Analysis of formalin-fixed, paraffin-embedded (FFPE) tissues from biopsy libraries has been, so far, a challenge for proteomics biomarker studies. To date, paraffin-embedded, formalin-fixed tissues are limited candidates for MALDI MS tissue profiling because these treatments may hamper efficient ionization of the proteins and peptides. There are recent methodological reports demonstrating the principal feasibility of MALDI-IMS on FFPE tissues (Stauber et al. 2008; Lemaire et al. 2007a; Wisztorski et al. 2007).
Selection and application of MALDI matrices
The multiple roles played by the matrix in the desorption and ionization of large analytes has lead to the optimum choice of matrices for different molecular classes being empirically determined (McDonnell et al. 2005). For tissue analysis, it has been reported that sinapinic acid provides the best signals for higher molecular weight proteins, whereas α-cyano-4-hydroxycinnamic acid is more suitable for lower molecular weight peptides (Schwartz et al. 2003). For higher spatial resolution analysis, in which deposition of the matrix solution has been carefully controlled to minimize spatial relocation, sinapinic acid has been recommended (Kruse and Sweedler 2003).
Matrix application protocols include manual methods such as spraying using an airbrush or TLC sprayer or dipping the tissue sections into matrix containing solutions (Schwartz et al. 2003). An intrinsic disadvantage of these manual procedures is poor reproducibility; higher reproducibility can be achieved using automated sample preparation devices. These devices fall into two classes: spotting devices and spraying devices. Matrix spotters (e.g., Labcyte Portrait, Shimadzu ChiP or Leaptec TMiD) apply small droplets of matrix solution in a grid onto the tissue. Underneath each droplet the spatial information is lost, so the lateral resolution is defined by the spot-to-spot distance. This distance can actually be significantly larger than the actual droplet size, because the requirement for spotting onto the same position repeatedly to achieve sufficient matrix coating causes significant loss of resolution due to small positioning inaccuracies. In addition, capillary forces cause some droplets moving on the tissue off their landing site. Another reason for reduced spotting resolution is crystallizing matrix at the nozzle of, e.g., piezoelectric dispensers which affects the angle of droplet impact. Robot designs without a nozzle can efficiently eliminate the clogging problem, though not the previously listed effects. Here the droplet is ejected from an open reservoir by a sonic impulse (Labcyte) (Aerni et al. 2006). Spotting robots can also be used to add chemical reagents onto the tissue in a controlled way to perform chemical modifications or digestions on the tissue, as reported with the CHiP (Shimadzu) (Groseclose et al. 2007). However, spotting robots suffer with regard to preparation time, which increases quadratically with the tissue area and targeted resolution, and lateral resolution limitations. Spotters are limited to provide for image resolutions in the 200–500 μm range. Because smaller droplets do not extract the analyte as efficient as large ones, the spectra quality rapidly drops if preparations for higher image resolutions are used. Significantly higher resolutions can be achieved by spray preparation of the matrix onto the tissue. Two spraying principles are currently utilized in commercial devices: pneumatic spray and vibrational spray. The ImagePrep device (Bruker Daltonics) utilizes vibrational vaporization of the matrix with a piezo-electric spray head. The spray head moves a pinhole sheet next to the matrix reservoir to eject small droplets with an average diameter of 20 μm. These droplets deposit onto the tissue and incubate in a controlled atmosphere chamber. The entire process of spraying and drying is monitored by an optical scattering-light sensor that allows the evaluation of the tissue wetness, matrix thickness and drying rate. Therefore, ImagePrep allows high resolutions of up to 25 μm with a good spectra quality by push button operation, being a significant advantage for routine operation. Another spray robot, the TM-sprayer (Leaptec), uses a heated capillary with a pneumatic spray that moves in predefined patterns over the tissue sample. This makes the spray more reproducible than a manual pneumatic spray. Parameters such as the drying rate and the wetness of the tissue are not monitored during the preparation but have to be tuned in advance.
Histological staining of tissues and MALDI-IMS
For the interpretation of MALDI-IMS results it is an absolute necessity to correlate the MALDI image with the histological information. Advanced MALDI-IMS software allows superimposing the MALDI images over a macroscopic or microscopic optical image of the sample taken before the MALDI measurement. While that primary macroscopic optical image is sufficient to recognize the outline of the tissue and to define the measurement area, it is usually not possible to see histological features in that image in contrast to microscopic images. For a histomorphological interpretation it is necessary to use stained tissue sections. Two approaches have been used to correlate histology with the MALDI-IMS result so far.
Histological staining of a consecutive section
In this approach, one section is used for the MALDI imaging, and another section is used to do the histological staining. This approach allows the use of any staining protocol, especially to use a Hematoxylin–Eosin (H&E) staining. This staining technique is a preferred histological staining because it yields a high degree of information on the sample. The main difficulty with this approach is the fact that the MALDI image is derived from a different section than the histological image. It may be sufficient to differentiate rather large tissue features, such as large invasive tumor regions from large connective tissue regions. To some degree it remains guesswork if the features seen in the histology are properly matched with the molecular information.
Histological staining of the section prior to the MALDI-IMS
This approach has been reported to integrate histology and MALDI-IMS (Chaurand et al. 2004b), and it does allow an unambiguous correlation of histomorphology and MALDI-IMS. One of the commonly used staining procedures in clinical pathology employs H&E, although unfortunately the quality of the mass spectra from these sections is significantly compromised relative to that obtained from unstained sections. To circumvent these limitations, protocols were developed to permit histomorphology and protein profiling to be performed on the very same tissue section. A series of commonly used histological dyes was tested for compatibility with mass spectrometric analysis. It was found that cresyl violet and methylene blue, as well as several other dyes, do not compromise overall mass spectra quality and allow specific regions of tissues to be easily analyzed. This approach has two disadvantages, however, first, it subjects the sample to additional handling steps prior the measurement. Second, and more severe is the fact that the choice of stains is limited to MALDI compatible ones. These stains (e.g., methylene blue or DAPI) are sufficient to localize cells in the histology, but they do not yield the same information as an H&E stain, so crucial information on the sample may be missed and it is still necessary to do a full H&E staining on a consecutive section.
Recently a new approach has been reported: The staining of the sample after the MALDI measurement (Schwamborn et al. 2007). This allows the use of H&E staining and an unambiguous correlation with the MALDI-IMS results. However, a prerequisite for this approach is the integrity of tissue under the MALDI measurement, shown in Fig. 1. Further MALDI-IMS experiments shown in the Figs. 2, 3, and 4 of this review are performed by staining of the sample after the MALDI measurement.

Integrity of histomorphology after MALDI measurement of tissue. a Frozen sample of a human pancreatic tissue on a cryostat steel plate. b Tissue section (10 μm) mounted onto a conductive glass slide for MALDI-IMS. c Tissue section after MALDI measurement: MALDI matrix was coated on the tissue section before measurement. d Subsequent to MALDI measurement the matrix is removed and the very same section is stained using standard H&E. e The histomorphology after MALDI-IMS is well-preserved as shown on higher magnification. Scale bars 4 mm (b–d) and 400 μm (e)

Comparison of non-cancerous (exocrine pancreas) and cancerous (invasive ductal pancreatic cancer) tissue by MALDI-IMS. a Histological image of the tissue section after MALDI-IMS. b Selected mass species correlates with non-cancerous (m/z 14,836) tissue area. c Selected mass species correlates with cancerous (m/z 13,777) tissue area. d Average spectra in the mass range of 12–16.5 kDa obtained from the non-cancerous (green) and the cancerous (red) region displaying the two masses visualized in (b) and (c). e Corresponding Pseudo-Gel view of (d). Scanning resolution 160 μm. Scale bars 2 mm

MALDI-IMS of a tissue section of rat pituitary gland. a Optical microscopic image of a H&E stained tissues section. The staining was done after the MALDI measurement of the tissue section. b–d Visualized selected m/z species representing features to pars distalis (m/z 6,651; green), pars intermedia (m/z 2,897; red) and pars neuralis (m/z 9,685; yellow). e Merge of a–d. f MALDI-TOF MS spectra obtained from this case from pars distalis (green), pars intermedia (red) and pars neuralis (yellow) showing molecular differences between the histological regions. Scanning resolution 50 μm. Scale bars 1 mm

Mass species representing molecular features of preinvasive and invasive lesions of the breast. a Optical microscopic image of a H&E stained tissues section showing several carcinomata in situ regions (outlined in green). The staining was done after the MALDI measurement of the tissue section. This allows an unambiguous correlation with the MALDI imaging results. b Visualization of ion density images of two selected masses (m/z = 9,750 shown in yellow, m/z = 4,519 shown in blue) c Overlay of H&E staining and molecular image. The distribution of these two masses suggests a different clonal evolution of the preinvasive lesions. These two masses are also present in the invasive cancer cells surrounding some carcinomata in situ (right site). Scanning resolution 80 μm. Scale bars 1 mm
Data processing, visualization and statistical analysis
In MALDI-IMS very information-rich datasets with hundreds of mass signals can be obtained. This is clearly an advantage, but it may become a limitation when a large number of datasets have to be evaluated peak-by-peak, because the evaluation can be very time consuming. Many tools have been developed to visualize and analyze this vast dataset intelligently and efficiently. Using different data processing strategies for visualization, it enables fast mass spectrometric imaging of large surfaces at high-spatial resolution and thus aids in the understanding of various diseases and disorders (Klinkert et al. 2007, Villmann et al. 2008). This includes many basic but widely used features, such as region-of-interest analysis, choice of intensity scales for image display (color palette, linear/logarithmic), image overlay, binning of spectral and image data for improved signal-to-noise, intensity profiles (variation of intensity with time or space), and chemical libraries for analyte identification (McDonnell and Heeren 2007). In addition to these facilities, imaging mass spectrometrists have continued to develop a broad array of tools. These have improved the quality of information extracted from the large data sets, reduced the dimensionality to more practical levels and allowed different imaging data sets to be aligned and compared. Several available software programs address data management, including determination of statistical significance and relative abundance between particular protein species. Moreover, the need for such software amid the growing number of proteomics laboratories will ensure that such programming will be standardized, continually upgraded, and user friendly. For clinical studies, large numbers of patients need to be studied, each having unique aspects to their disease. Indeed, the base premise of individualized medicine is founded on this exciting but enormously complex molecular diversity. It is evident that innovative biocomputational studies are an absolute necessity in identifying individualized molecular patterns to aid in diagnosis and prognosis. Examples of data analysis algorithms have been published and consist of four steps. Proteins are selected that are differentially expressed among histomorphological defined groups. This selection is based on the Kruskal–Wallis test, Fisher’s exact test (dichotomizing the expression level as present or not), the Student’s t test, significance analysis of microarrays, weighted gene analysis, and the modified info score methods. The cutoff points for each method are P < 0.0001, P < 0.0001, P < 0.0001, 3.5, 2, and 0, respectively. Proteins are included in the final list if they meet at least three of these six selection criteria (Caldwell and Caprioli 2005). One technique to reduce the complexity of the information in multidimensional datasets in MALDI-IMS is the principal component analysis (PCA) (Van de Plas et al. 2007). This is a transformation of the original coordinate system defined by peak intensities to a coordinate system that better explains the variance in the dataset. The new coordinates are called principal components and are ordered in decreasing number of variance. By removing those PC’s that do not contain a lot of information the dimensionality of the dataset can be reduced to a large extent. The principal component scores can then be used to reconstruct a few images that contain most of the information.
While the PCA reduces the dimensionality of the dataset, it does not classify the spectra. An unsupervised classification can be achieved by hierarchical clustering (McCombie et al. 2005). Here the spectra are pair-wise clustered according to similarity until a dendrogram is obtained that contains all spectra. Each branch of the dendrogram can be considered a class of spectra. It is now possible for a tissue expert to select those dendrogram nodes that reflect certain histological features. The flexImaging software, e.g., provides a interactive dendrogram browser in which dendrogram nodes can be selected and the spectra that belong to that node are displayed on the image. Several branches can be selected with different colors to reconstruct a full image. This makes the evaluation of complex datasets much faster than evaluating hundreds of individual mass signals. An example of hierarchical clustering of a mouse kidney dataset is shown in Fig. 5. Further examples illustrate the applications of various software tools for data analysis (Figs. 2, 3, 4, 6).

Hierarchical Clustering of a mouse kidney dataset achieved by MALDI-IMS. a Full dendrogram of all spectra in a mouse kidney dataset. b Optical image of the mouse kidney analysed by MALDI-IMS. c and d Reconstruction of selected dendrogram branches and corresponding images. The three main branches reflect the renal cortex (blue), medulla (green) and pelvis (red) c The medulla branch separates into two distinct areas, while cortex branch further differentiates into fat and connective tissue of the renal capsule and hylus and the actual cortex (d)

Histology directed MALDI-IMS profiling of gastric mucosa and esophageal squamous epithelium. a Overall average spectra in the mass range of 4.3–7.4 kDa obtained from eight cases of human gastric mucosa (blue) and eleven cases of esophageal squamous epithelium (green). b Virtual gel view of individual spectra displayed for mass range of 4.3–7.4 kDa. Statistical analysis revealed in this experiment 63 differentially expressed m/z species at significant levels
Protein identification
Identification of differentially expressed peptides and proteins in tissues enhances the understanding of the biological processes underlying disease. After processing of the raw mass spectra, and determination of statistical significance, a list of spectral features (m/z species) is generated. These m/z species represent peptides and proteins with expressions that are significantly modulated by the disease. The identification strategy may vary for a particular protein because of sample complexity, protein abundance, or molecular weight (Fig. 7). There are two approaches to protein identification. The top–down approach involves ionization and gas phase fragmentation of the protein of interest inside the mass spectrometer (Reid and McLuckey 2002), whereas the bottom–up approach utilizes MS to identify peptides obtained from protease digestion of that protein, often in a mixture of other proteolytic fragments (Wysocki et al. 2005). The resulting mass spectra are searched against theoretical protein/peptide databases for corresponding sequence patterns. Searches are performed using conventional algorithms such as MASCOT and Sequest (Hirosawa et al. 1993; Link et al. 1999).

Workflow of protein identification
Recent work has shown that digestion and identification of proteins may be coupled with direct tissue analysis (Groseclose et al. 2007). This technique involves automatically depositing a spotted array of enzymatic solution onto the tissue at room temperature. After hydrolysis, MALDI matrix is deposited onto the array for subsequent MALDI MS analysis. The in situ identification of proteins requires less time than conventional protein identification strategies and the ability to compare a given protein image to the image of its subsequent peptides increases identification confidence. This process is best used for validation, i.e., where the presence of one or more specific proteins is needed to be established (Herring et al. 2007).
Finally, immunohistochemistry can be employed to verify protein identification, assuming appropriate antibodies for the protein exist.
Application of MALDI-IMS to tissue analysis
A majority of MALDI-IMS studies have been dedicated to the study of proteins contained in human and animal tissue sections (Cornett et al. 2007). Applications in the field of pathology hold particular interest for many because of the potential benefit for clinical diagnoses and treatment. These applications are complemented by the field of MALDI-IMS for the detection of drugs and metabolites in tissues. Other studies have focused on proteomic events occurring in normally developing tissues.
Diagnostic and prognostic assessments in clinical pathology
To date, profiling and imaging by MALDI-IMS has been applied to multiple diseased tissues, including human non-small-cell lung tumors (Yanagisawa et al. 2003), gliomas (Schwartz et al. 2005), breast cancer (Cornett et al. 2006) and ovarian tumors (Lemaire et al. 2007b) and continues to be a main focus of MALDI-IMS studies (Chaurand et al. 2004a; Caprioli 2005; Caldwell and Caprioli 2005; Sköld et al. 2006; Stoeckli et al. 2007; Meistermann et al. 2006; Cornett et al. 2006; Herring et al. 2007; Corbin et al. 2008). Molecular protein signatures represent a unique data set with which to classify and correlate clinically relevant information and outcomes with changing molecular events ongoing in the progression and treatment of disease. The general approach taken by these studies is comparative proteomic analyses whereby mass spectral features (m/z peaks) are correlated with a variety of patient data, such as therapeutic regimen and overall outcome. In some cases, the goal of the experiment is to identify specific molecular changes associated with progression of disease. For example, molecular signatures from glioma or non-small-cell lung cancer can be used to distinguish tumor grade and were found to be correlated with patient survival (Yanagisawa et al. 2003; Schwartz et al. 2005). Identification of specific cellular changes within heterogeneous tissue can be particularly challenging from the perspective of matrix spot size, placement and lateral resolution (Cornett et al. 2007). From work already published, it seems likely that the integration of this technology into protocols for disease diagnosis as well as outcome prediction will soon take place. As protein expression data becomes available from various tissue types, this approach will provide a common disease-wide approach that can be applied to many specific problems. One can envision that, e.g., the use of MALDI-IMS technology to evaluate a “tumour of unknown primary.” Current data suggests that MALDI MS might be superior to immunohistochemical stains in identifying the site of origin for such tumors (Chaurand et al. 2004a). The potential capability of MALDI-IMS to measure response to therapeutic agents in tumor and surrounding tissues is a particularly exciting application of this technology. The original protein profile obtained from the primary tumor could be used to influence the selection of therapeutic agents. Levels of drugs such as chemotherapeutic agents could be measured directly from a tissue biopsy to assess adequacy of delivery to a particular organ site. The ability of drugs and other bioreactive molecules to adequately penetrate larger tumors has long been known to be problematic and could be more adequately assessed by this technology. In addition, alterations in specific molecular pathways directly modulated or indirectly affected by the agent could be evaluated. Studies of this type that establish proof of principle have been reported (Reyzer et al. 2003). Similar methods could be envisioned to monitor patients treated with conservative therapy for relapse. Together with genomics and perhaps additional molecular information describing the state of lipids and metabolites, proteomics provides an entry into individualized medicine, where each patient’s disease is unique at the molecular level and will be treated accordingly (Chaurand et al. 2004a).
Small-molecule imaging and drug metabolism
During drug discovery often the question is raised as to whether the drug can reach the site of action which helps researchers better assess the potential value of that compound as a pharmaceutical product and toxicological outcomes (Hsieh et al. 2007). MALDI-IMS opens the possibility to directly determine the distribution of pharmaceuticals in tissue sections which might unravel their disposition or biotransformation pathway for new drug development. Many compounds that are biologically or pharmacologically relevant are less than 1 kDa in size and thus fall into the broad category of small molecules (Cornett et al. 2007). These include both exogenous and endogenous molecules, such as pharmaceutical compounds and their metabolites, drugs of abuse, environmental toxins, endogenous metabolites and lipids. Compared to the standard method in this field, the whole-body autoradiography (WBA), MALDI imaging offers some advantages but also some drawbacks. In WBA, a radioactive labeled drug is administered to the animal. Thin sections of the animals are then exposed to photographic film which detects the radioactivity. The advantages of MALDI-IMS in contrast to WBA are that it is not needed to use radiolabeled drugs. This is a big advantage especially in early stages of drug development since there large numbers of compounds need to be screened and preparing radiolabeled drugs is tedious. The second advantage of MALDI-IMS is that it offers the possibility to differentiate between the drug and its metabolites, while in WBA the radioactivity may come from either drug or metabolite. On the other hand, WBA will always yield a result and has a very low detection limit. In MALDI-IMS, there is always a possibility that the ion of interest is suppressed in desorption. The detection limit of MALDI-IMS is strongly dependent on the drug, some drugs may not even be seen. This is why MALDI-IMS of drugs has found its main application in drug safety studies, where high doses of drugs are used. Drug imaging differs from other MALDI imaging applications such a way that the compound to be detected is known in advance. The mass range in which the drugs are measured is usually crowded by a large number of signals that come from the MALDI matrix as well as from lipids and naturally occurring substances. It is therefore necessary to ensure the necessary specificity of detection of a particular, probably small signal in between many other signals. This has been so far mainly achieved by using a MS/MS measurement. The ion of interest is selected and fragmented. The quantification and reconstruction of the MALDI image is then done on a specific fragment of the drug. Interfering signals of similar mass in MS mode usually yield different MS/MS signals, so the necessary specificity of the measurement is achieved. Recently MALDI-IMS became available on instruments, that allow the measurement of mass signals with very high resolution and precision, namely Fourier-transform mass spectrometers (Cornett et al. 2008). In these instruments the necessary specificity of the drug imaging experiment can be reached by measuring the precise mass of the drug. Matrix and background ions that would interfere in instruments with lower resolution can be separated in the MS mode alone. Applications of small-molecule MALDI-IMS focused on pharmaceutically active compounds and their metabolites (Khatib-Shahidi et al. 2006; Hsieh et al. 2006, 2007), and considerable effort continues in this field (Reyzer and Caprioli 2007). For example, one study reported the localization of the drug olanzapine and two-first-pass metabolites in whole-rat sections (Khatib-Shahidi et al. 2006). The results of this study correlated well with both autoradiography and LC-MS/MS quantitative results, illustrating the power of MALDI-IMS for metabolite profiling. More recently, there has been considerable interest in adding quantitative capabilities to the MALDI-IMS experiment (Signor et al. 2007; Corr et al. 2006). The technology also opens up the exciting possibility of correlating drug distribution with concomitant protein changes (Reyzer et al. 2004).
Developmental biology
Protein imaging and profiling using MALDI-IMS has become a powerful method for analyzing changes in global protein expression patterns in cells and tissues as a function of developmental processes (Cornett et al. 2007). Proteomic studies using MALDI-IMS can yield insight into some complex biological processes. MALDI-IMS analyses of epididymal sections have been used to determine the localized composition of the epididymal fluid from caput to cauda (Chaurand et al. 2003), confirming that different protein compositions are found along the length of the epididymis. Other recent examples include the direct imaging of secretory peptides from frog skin (Brand et al. 2006) and bovine and rabbit ocular lens proteins (Han and Schey 2006; Grey and Schey 2008), as well as neuropeptides from rat pituitary (Altelaar et al. 2007), decapod crustacean neuronal tissue (DeKeyser et al. 2007), protein profiles during embryo implantation (Burnum et al. 2008) or mouse prostate development (Chaurand et al. 2008).
Imaging of phospholipids
Lipidomics is one new frontier in biomarker research. Not only are lipids the main components in membranes that define the contours of the cell and its organelles, but they are also used for storage. Lipids form stable noncovalent complexes with proteins as well as with many drugs. Lipids are a storage depot for drugs and certain types of organic molecules. Analysis of lipids is challenging because of the wide molecular diversity of this class of compounds and their relative insolubility in aqueous systems. Direct tissue imaging by MALDI-IMS is a powerful tool as it gives a more complete and accurate structural picture and can trace and follow where drugs localize in tissue with minimal anatomical disruption and a minimum of manipulations. The use of ion mobility has also been reported in conjunction with MALDI MS as a means to fractionate lipids (Woods and Jackson 2006; Jackson et al. 2005a, b; Cornett et al. 2007; McLean et al. 2007). In this technique, ions are first separated by ion mobility and then analyzed by TOF mass spectrometry. The desorbed lipid ions fall on a trend line (a function of charge and collision cross-section) that is separate from those of oligonucleosides, peptides, proteins, and drugs and metabolites having the same nominal mass. This allows ions originating from lipids to be distinguished from other small molecules. Another study reported label-free imaging mass spectrometry to study lipid–lipid interactions in a model membrane system (Zheng et al. 2007).
Conclusions
The emergence of MALDI-IMS has provided an enormous impulse to the field of imaging mass spectrometry. Direct tissue analysis by MALDI-IMS is an important technology for assessing the localization of molecular species and for revealing the underlying molecular signatures indicative of disease. The molecular species identified in these experiments can provide insight into mechanisms and etiology of disease. It is now commercially available and is widely used to record the spatial distributions of numerous peptide and protein ions, in parallel and without a label, from tissue sections and cells. It has been applied to study the changes in protein expression levels and distributions associated with a range of pathologies. Furthermore, the same techniques are used to trace the distribution of pharmaceuticals and their metabolites within organs and complete animals, as well as to investigate the resulting changes in the organ’s proteome. The potential of MALDI imaging has driven many developments covering all aspects of the experiment. Advances in data acquisition now allow fast MALDI imaging with high-spatial resolution. Developments in sample preparation have also continued unabated. Many of the developments discussed in this review can be combined, the results can then be analyzed using multivariate techniques or combined with information from other imaging techniques using cross correlation analysis (Sinha et al. 2008). These results can then be compared with the results of global proteomics/lipidomics strategies applied to spatially resolved samples, either through voxelization, laser-capture microdissection, or any other sample preparation protocol. It is this potential, which is slowly being fulfilled, to obtain extensive spatially resolved biomolecular information, an absolute necessity for a thorough picture of the molecular processes underpinning biological and pathological processes or to track the distribution of pharmaceuticals that is attracting ever more interest.
References
Aebersold R, Goodlett DR (2001) Mass spectrometry in proteomics. Chem Rev 101(2):269–295
Aerni HR, Cornett DS, Caprioli RM (2006) Automated acoustic matrix deposition for MALDI sample preparation. Anal Chem 78(3):827–834
Altelaar AF, van Minnen J, Jiménez CR, Heeren RM, Piersma SR (2005) Direct molecular imaging of Lymnaea stagnalis nervous tissue at subcellular spatial resolution by mass spectrometry. Anal Chem 77(3):735–741
Altelaar AF, Klinkert I, Jalink K, de Lange RP, Adan RA, Heeren RM, Piersma SR (2006) Gold-enhanced biomolecular surface imaging of cells and tissue by SIMS and MALDI mass spectrometry. Anal Chem 78(3):734–742
Altelaar AF, Luxembourg SL, McDonnell LA, Piersma SR, Heeren RM (2007) Imaging mass spectrometry at cellular length scales. Nat Protoc 2(5):1185–1196
Bhattacharya SH, Gal AA, Murray KK (2003) Laser capture microdissection MALDI for direct analysis of archival tissue. J Proteome Res 2(1):95–98
Brand GD, Krause FC, Silva LP, Leite JR, Melo JA, Prates MV, Pesquero JB, Santos EL, Nakaie CR, Costa-Neto CM, Bloch C Jr (2006) Bradykinin-related peptides from Phyllomedusa hypochondrialis. Peptides 27(9):2137–2146
Burnum KE, Tranguch S, Mi D, Daikoku T, Dey SK, Caprioli RM (2008) Imaging mass spectrometry reveals unique protein profiles during embryo implantation. Endocrinology. 2008 Apr 10
Caldwell RL, Caprioli RM (2005) Tissue profiling by mass spectrometry: a review of methodology and applications. Mol Cell Proteomics 4(4):394–401
Caprioli RM (2005) Deciphering protein molecular signatures in cancer tissues to aid in diagnosis, prognosis, and therapy. Cancer Res 65(23):10642–10645
Caprioli RM, Farmer TB, Gile J (1997) Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anal Chem 69(23):4751–4760
Chaurand P, Caprioli RM (2002) Direct profiling and imaging of peptides and proteins from mammalian cells and tissue sections by mass spectrometry. Electrophoresis 23(18):3125–3135
Chaurand P, Stoeckli M, Caprioli RM (1999) Direct profiling of proteins in biological tissue sections by MALDI mass spectrometry. Anal Chem 71(23):5263–5270
Chaurand P, DaGue BB, Pearsall RS, Threadgill DW, Caprioli RM (2001) Profiling proteins from azoxymethane-induced colon tumors at the molecular level by matrix-assisted laser desorption/ionization mass spectrometry. Proteomics 1(10):1320–1326
Chaurand P, Schwartz SA, Caprioli RM (2002) Imaging mass spectrometry: a new tool to investigate the spatial organization of peptides and proteins in mammalian tissue sections. Curr Opin Chem Biol 6(5):676–681
Chaurand P, Fouchécourt S, DaGue BB, Xu BJ, Reyzer ML, Orgebin-Crist MC, Caprioli RM (2003) Profiling and imaging proteins in the mouse epididymis by imaging mass spectrometry. Proteomics 3(11):2221–2239
Chaurand P, Sanders ME, Jensen RA, Caprioli RM (2004a) Proteomics in diagnostic pathology: profiling and imaging proteins directly in tissue sections. Am J Pathol 165(4):1057–1068
Chaurand P, Schwartz SA, Billheimer D, Xu BJ, Crecelius A, Caprioli RM (2004b) Integrating histology and imaging mass spectrometry. Anal Chem 76(4):1145–1155
Chaurand P, Schwartz SA, Reyzer ML, Caprioli RM (2005) Imaging mass spectrometry: principles and potentials. Toxicol Pathol 33(1):92–101
Chaurand P, Rahman MA, Hunt T, Mobley JA, Gu G, Latham JC, Caprioli RM, Kasper S (2008) Monitoring mouse prostate development by profiling and imaging mass spectrometry. Mol Cell Proteomics 7(2):411–423
Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319(5865):962–965
Cornett DS, Mobley JA, Dias EC, Andersson M, Arteaga CL, Sanders ME, Caprioli RM (2006) A novel histology-directed strategy for MALDI-MS tissue profiling that improves throughput and cellular specificity in human breast cancer. Mol Cell Proteomics 5(10):1975–1983
Cornett DS, Reyzer ML, Chaurand P, Caprioli RM (2007) MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat Methods 4(10):828–833
Cornett DS, Frappier SL, Caprioli RM (2008) MALDI-FTICR imaging mass spectrometry of drugs and metabolites in tissue. Anal Chem [Epub ahead of print]
Corr JJ, Kovarik P, Schneider BB, Hendrikse J, Loboda A, Covey TR (2006) Design considerations for high speed quantitative mass spectrometry with MALDI ionization. J Am Soc Mass Spectrom 17(8):1129–1141
Curran S, McKay JA, McLeod HL, Murray GI (2000) Laser capture microscopy. Mol Pathol 53(2):64–68
DeKeyser SS, Kutz-Naber KK, Schmidt JJ, Barrett-Wilt GA, Li L (2007) Imaging mass spectrometry of neuropeptides in decapod crustacean neuronal tissues. J Proteome Res 6(5):1782–1791
Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM (1989) Electrospray ionization for mass spectrometry of large biomolecules. Science 246(4926):64–71
Godovac-Zimmermann J, Brown LR (2001) Perspectives for mass spectrometry and functional proteomics. Mass Spectrom Rev 20(1):1–57
Grey AC, Schey KL (2008) Distribution of bovine and rabbit lens alpha-crystallin products by MALDI imaging mass spectrometry. Mol Vis 14:171–179
Groseclose MR, Andersson M, Hardesty WM, Caprioli RM (2007) Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J Mass Spectrom 42(2):254–262
Han J, Schey KL (2006) MALDI tissue imaging of ocular lens alpha-crystallin. Invest Ophthalmol Vis Sci 47(7):2990–2996
Heeren RM (2005) Proteome imaging: a closer look at life’s organization. Proteomics 5(17):4316–4326
Herring KD, Oppenheimer SR, Caprioli RM (2007) Direct tissue analysis by matrix-assisted laser desorption ionization mass spectrometry: application to kidney biology. Semin Nephrol 27(6):597–608
Hillenkamp F, Karas M, Beavis RC, Chait BT (1991) Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal Chem 63(24):1193A–1203A
Hirosawa M, Hoshida M, Ishikawa M, Toya T (1993) MASCOT: multiple alignment system for protein sequences based on three-way dynamic programming. Comput Appl Biosci 9:161–167
Hsieh Y, Casale R, Fukuda E, Chen J, Knemeyer I, Wingate J, Morrison R, Korfmacher W (2006) Matrix-assisted laser desorption/ionization imaging mass spectrometry for direct measurement of clozapine in rat brain tissue. Rapid Commun Mass Spectrom 20(6):965–972
Hsieh Y, Chen J, Korfmacher WA (2007) Mapping pharmaceuticals in tissues using MALDI imaging mass spectrometry. J Pharmacol Toxicol Methods 55(2):193–200
Jackson SN, Wang HY, Woods AS (2005a) Direct profiling of lipid distribution in brain tissue using MALDI-TOFMS. Anal Chem 77(14):4523–4527
Jackson SN, Wang HY, Woods AS, Ugarov M, Egan T, Schultz JA (2005b) Direct tissue analysis of phospholipids in rat brain using MALDI-TOFMS and MALDI-ion mobility-TOFMS. J Am Soc Mass Spectrom 16(2):133–138
Karas M, Hillenkamp F (1988) Laser desorption ionization of proteins with molecular masses exceeding 10, 000 daltons. Anal Chem 60(20):2299–2301
Khatib-Shahidi S, Andersson M, Herman JL, Gillespie TA, Caprioli RM (2006) Direct molecular analysis of whole-body animal tissue sections by imaging MALDI mass spectrometry. Anal Chem 78(18):6448–6456
Klinkert I, McDonnell LA, Luxembourg SL, Altelaar AF, Amstalden ER, Piersma SR, Heeren RM (2007) Tools and strategies for visualization of large image data sets in high-resolution imaging mass spectrometry. Rev Sci Instrum 78(5):053716
Kruse R, Sweedler JV (2003) Spatial profiling invertebrate ganglia using MALDI MS. J Am Soc Mass Spectrom 14(7):752–759
Lahm HW, Langen H (2000) Mass spectrometry: a tool for the identification of proteins separated by gels. Electrophoresis 21(11):2105–2114
Lavoie C, Paiement J (2008) Topology of molecular machines of the endoplasmic reticulum: a compilation of proteomics and cytological data. Histochem Cell Biol 129(2):117–128
Lemaire R, Desmons A, Tabet JC, Day R, Salzet M, Fournier I (2007a) Direct analysis and MALDI imaging of formalin-fixed, paraffin-embedded tissue sections. J Proteome Res 6(4):1295–1305
Lemaire R, Menguellet SA, Stauber J, Marchaudon V, Lucot JP, Collinet P, Farine MO, Vinatier D, Day R, Ducoroy P, Salzet M, Fournier I (2007b) Specific MALDI imaging and profiling for biomarker hunting and validation: fragment of the 11S proteasome activator complex, Reg alpha fragment, is a new potential ovary cancer biomarker. J Proteome Res 6(11):4127–4134
Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR 3rd (1999) Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol 17:676–682
Masumori N, Thomas TZ, Chaurand P, Case T, Paul M, Kasper S, Caprioli RM, Tsukamoto T, Shappell SB, Matusik RJ (2001) A probasin-large T antigen transgenic mouse line develops prostate adenocarcinoma and neuroendocrine carcinoma with metastatic potential. Cancer Res 61(5):2239–2249
McCombie G, Staab D, Stoeckli M, Knochenmuss R (2005) Spatial and spectral correlations in MALDI mass spectrometry images by clustering and multivariate analysis. Anal Chem 77(19):6118–6124
McDonald WH, Yates JR 3rd (2000) Proteomic tools for cell biology. Traffic 1(10):747–754
McDonnell LA, Heeren RM (2007) Imaging mass spectrometry. Mass Spectrom Rev 26(4):606–643
McDonnell LA, Piersma SR, MaartenAltelaar AF, Mize TH, Luxembourg SL, Verhaert PD, van Minnen J, Heeren RM (2005) Subcellular imaging mass spectrometry of brain tissue. J Mass Spectrom 40(2):160–168
McLean JA, Ridenour WB, Caprioli RM (2007) Profiling and imaging of tissues by imaging ion mobility-mass spectrometry. J Mass Spectrom 42(8):1099–1105
Meistermann H, Norris JL, Aerni HR, Cornett DS, Friedlein A, Erskine AR, Augustin A, De Vera Mudry MC, Ruepp S, Suter L, Langen H, Caprioli RM, Ducret A (2006) Biomarker discovery by imaging mass spectrometry: transthyretin is a biomarker for gentamicin-induced nephrotoxicity in rat. Mol Cell Proteomics 5(10):1876–1886
Palmer-Toy DE, Sarracino DA, Sgroi D, LeVangie R, Leopold PE (2000) Direct acquisition of matrix-assisted laser desorption/ionization time-of-flight mass spectra from laser capture microdissected tissues. Clin Chem 46(9):1513–1516
Pandey A, Mann M (2000) Proteomics to study genes and genomes. Nature 405(6788):837–846
Pierson J, Norris JL, Aerni HR, Svenningsson P, Caprioli RM, Andrén PE (2004) Molecular profiling of experimental Parkinson’s disease: direct analysis of peptides and proteins on brain tissue sections by MALDI mass spectrometry. J Proteome Res 3(2):289–295
Reid GE, McLuckey SA (2002) ‘Top down’ protein characterization via tandem mass spectrometry. J Mass Spectrom 37:663–675
Reyzer ML, Caprioli RM (2007) MALDI-MS-based imaging of small molecules and proteins in tissues. Curr Opin Chem Biol 11(1):29–35
Reyzer ML, Hsieh Y, Ng K, Korfmacher WA, Caprioli RM (2003) Direct analysis of drug candidates in tissue by matrix-assisted laser desorption/ionization mass spectrometry. J Mass Spectrom 38(10):1081–1092
Reyzer ML, Caldwell RL, Dugger TC, Forbes JT, Ritter CA, Guix M, Arteaga CL, Caprioli RM (2004) Early changes in protein expression detected by mass spectrometry predict tumor response to molecular therapeutics. Cancer Res 64(24):9093–9100
Roepstorff P (1997) Mass spectrometry in protein studies from genome to function. Curr Opin Biotechnol 8(1):6–13
Rohner TC, Staab D, Stoeckli M (2005) MALDI mass spectrometric imaging of biological tissue sections. Mech Ageing Dev 126(1):177–185
Rujoi M, Estrada R, Yappert MC (2004) In situ MALDI-TOF MS regional analysis of neutral phospholipids in lens tissue. Anal Chem 76(6):1657–1663
Schwamborn K, Krieg RC, Reska M, Jakse G, Knuechel R, Wellmann A (2007) Identifying prostate carcinoma by MALDI-Imaging. Int J Mol Med 20(2):155–159
Schwartz SA, Reyzer ML, Caprioli RM (2003) Direct tissue analysis using matrix-assisted laser desorption/ionization mass spectrometry: practical aspects of sample preparation. J Mass Spectrom 38(7):699–708
Schwartz SA, Weil RJ, Johnson MD, Toms SA, Caprioli RM (2004) Protein profiling in brain tumors using mass spectrometry: feasibility of a new technique for the analysis of protein expression. Clin Cancer Res 10(3):981–987
Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, Johnson MD, Caprioli RM (2005) Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Res 65(17):7674–7681
Shimma S, Sugiura Y, Hayasaka T, Zaima N, Matsumoto M, Setou M (2008) Mass imaging and identification of biomolecules with MALDI-QIT-TOF-based system. Anal Chem 80(3):878–885
Signor L, Varesio E, Staack RF, Starke V, Richter WF, Hopfgartner G (2007) Analysis of erlotinib and its metabolites in rat tissue sections by MALDI quadrupole time-of-flight mass spectrometry. J Mass Spectrom 42(7):900–909
Sinha TK, Khatib-Shahidi S, Yankeelov TE, Mapara K, Ehtesham M, Cornett DS, Dawant BM, Caprioli RM, Gore JC (2008) Integrating spatially resolved three-dimensional MALDI IMS with in vivo magnetic resonance imaging. Nat Methods 5(1):57–59
Sköld K, Svensson M, Nilsson A, Zhang X, Nydahl K, Caprioli RM, Svenningsson P, Andrén PE (2006) Decreased striatal levels of PEP-19 following MPTP lesion in the mouse. J Proteome Res 5(2):262–269
Stauber J, Lemaire R, Franck J, Bonnel D, Croix D, Day R, Wisztorski M, Fournier I, Salzet M (2008) MALDI imaging of formalin-fixed paraffin-embedded tissues: application to model animals of Parkinson disease for biomarker hunting. J Proteome Res 7(3):969–978
Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM (2001) Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat Med 7(4):493–496
Stoeckli M, Staab D, Staufenbiel M, Wiederhold KH, Signor L (2002) Molecular imaging of amyloid beta peptides in mouse brain sections using mass spectrometry. Anal Biochem 311(1):33–39
Stoeckli M, Staab D, Schweitzer A, Gardiner J, Seebach D (2007) Imaging of a beta-peptide distribution in whole-body mice sections by MALDI mass spectrometry. J Am Soc Mass Spectrom 18(11):1921–1924
Todd PJ, Schaaff TG, Chaurand P, Caprioli RM (2001) Organic ion imaging of biological tissue with secondary ion mass spectrometry and matrix-assisted laser desorption/ionization. J Mass Spectrom 36(4):355–369
Van de Plas R, Ojeda F, Dewil M, Van Den Bosch L, De Moor B, Waelkens E (2007) Prospective exploration of biochemical tissue composition via imaging mass spectrometry guided by principal component analysis. Pac Symp Biocomput 2007:458–469
Villmann T, Schleif FM, Kostrzewa M, Walch A, Hammer B (2008) Classification of mass-spectrometric data in clinical proteomics using learning vector quantization methods. Brief Bioinform 9(2):129–143
Wisztorski M, Lemaire R, Stauber J, Menguelet SA, Croix D, Mathé OJ, Day R, Salzet M, Fournier I (2007) New developments in MALDI imaging for pathology proteomic studies. Curr Pharm Des 13(32):3317–3324
Woods AS, Jackson SN (2006) Brain tissue lipidomics: direct probing using matrix-assisted laser desorption/ionization mass spectrometry. AAPS J 8(2):E391–E395
Wysocki VH, Resing KA, Zhang Q, Cheng G (2005) Mass spectrometry of peptides and proteins. Methods 35:211–222
Xu BJ, Caprioli RM, Sanders ME, Jensen RA (2002) Direct analysis of laser capture microdissected cells by MALDI mass spectrometry. J Am Soc Mass Spectrom 13(11):1292–1297
Yanagisawa K, Shyr Y, Xu BJ, Massion PP, Larsen PH, White BC, Roberts JR, Edgerton M, Gonzalez A, Nadaf S, Moore JH, Caprioli RM, Carbone DP (2003) Proteomic patterns of tumour subsets in non-small-cell lung cancer. Lancet 362(9382):433–439
Zheng L, McQuaw CM, Ewing AG, Winograd N (2007) Sphingomyelin/phosphatidylcholine and cholesterol interactions studied by imaging mass spectrometry. J Am Chem Soc 129(51):15730–15731
Acknowledgments
We thank Ulrike Buchholz, Claudia-Mareike Pflüger, Eleonore Samson, and Andreas Voss for excellent technical assistance in performing MALDI-IMS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Walch, A., Rauser, S., Deininger, SO. et al. MALDI imaging mass spectrometry for direct tissue analysis: a new frontier for molecular histology. Histochem Cell Biol 130, 421–434 (2008). https://doi.org/10.1007/s00418-008-0469-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0469-9