



Molecular and clinical diseasome of comorbidities in exacerbated COPD patients
- Rosa Faner1,2,8,
- Alba Gutiérrez-Sacristán3,8,
- Ady Castro-Acosta2,4,
- Solène Grosdidier3,
- Wenqi Gan5,
- Milagros Sánchez-Mayor3,
- Jose Luis Lopez-Campos2,6,
- Francisco Pozo-Rodriguez2,4,
- Ferran Sanz3,
- David Mannino5,
- Laura I. Furlong3 and
- Alvar Agusti1,2,7⇑
- 1Fundació Privada Clinic per a la Recerca Biomèdica, Barcelona, Spain
- 2CIBER Enfermedades Respiratorias (CIBERES), Spain
- 3Integrative Biomedical Informatics Group, Research Program on Biomedical Informatics (GRIB), Hospital del Mar Medical Research Institute (IMIM), Universitat Pompeu Fabra, Barcelona, Spain
- 4Instituto de Investigación, Hospital 12 de Octubre, Madrid, Spain
- 5Dept of Preventive Medicine and Environmental Health, University of Kentucky College of Public Health, Lexington, KY, USA
- 6Unidad Médico-Quirúrgica de Enfermedades Respiratorias, Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocio/Universidad de Sevilla, Sevilla, Spain
- 7Thorax Institute, Hospital Clinic, IDIBAPS, Univ. Barcelona, Barcelona, Spain
- 8Co-primary authors
- Alvar Agustí, Institut del Tòrax, Hospital Clínic, Villarroel 170, Escala 3, Planta 5, 08036 Barcelona, Spain. E-mail: alvar.agusti{at}clinic.ub.es
Abstract
The frequent occurrence of comorbidities in patients with chronic obstructive pulmonary disease (COPD) suggests that they may share pathobiological processes and/or risk factors.
To explore these possibilities we compared the clinical diseasome and the molecular diseasome of 5447 COPD patients hospitalised because of an exacerbation of the disease. The clinical diseasome is a network representation of the relationships between diseases, in which diseases are connected if they co-occur more than expected at random; in the molecular diseasome, diseases are linked if they share associated genes or interaction between proteins.
The results showed that about half of the disease pairs identified in the clinical diseasome had a biological counterpart in the molecular diseasome, particularly those related to inflammation and vascular tone regulation. Interestingly, the clinical diseasome of these patients appears independent of age, cumulative smoking exposure or severity of airflow limitation.
These results support the existence of shared molecular mechanisms among comorbidities in COPD.
Abstract
Half of the comorbidities observed in COPD patients hospitalised by an exacerbation share common molecular mechanisms http://ow.ly/PDiGg
Introduction
Patients with chronic obstructive pulmonary disease (COPD) often suffer comorbid diseases that deteriorate their health status, increase their risk of COPD exacerbation and worsen their prognosis [1–5]. It is unclear if comorbidities are causally related to COPD and/or share molecular pathways or risk factors (such as ageing, smoking and/or inactivity), the so-called shared component hypothesis [6–10]. Very recently, Divo et al. [11] used network analysis to explore the association between multiple comorbidities in 1969 patients with clinically stable COPD [12]. The results showed that the 79 comorbidities considered in the study, as well as several demographic (age, body mass index), clinical (level of dyspnoea) and functional characteristics (degree of airflow limitation, exercise capacity) also included in the analysis, were significantly interlinked and formed a complex network in which six modules (or sub-networks) could be identified [11, 12]. From these observations, authors suggested (but did not explore) that these comorbidities and clinical characteristics may share pathobiological processes (the shared component hypothesis) [11, 12]. Besides, whether or not such potentially shared molecular processes were causally linked to COPD and/or resulted from shared risk factors, such as ageing, smoking and/or inactivity, was also unclear [7, 9, 10].
We recently used a novel, unbiased, integrative network analysis approach to explore the biological relationships between COPD, its comorbidities and the chemical products contained in tobacco smoke [13]. We found, first, that comorbid diseases indeed shared genes, proteins and biological pathways with COPD and, secondly, that many of these molecules were actually targets for the chemicals contained in the tobacco smoke exposome [13]. This analysis was based on data-mining of published results, not on real-life clinical data, and potentially shared molecular commonalities between comorbidities themselves were not analysed. To address these limitations, here we sought to investigate the shared molecular basis across comorbidities in patients hospitalised because of an episode of COPD exacerbation, as well as the influence of age, cumulative smoking exposure and severity of airflow limitation. To this end, we used two convenient and large (n=5447), nationwide [14–17] databases of COPD exacerbation and a network analysis strategy [6, 7, 13, 18] where we compared their clinical diseasome with their corresponding molecular diseasome. The clinical diseasome is a network representation of relationships between comorbidities, in which diseases are connected if their co-occurrence is higher than that expected at random [18]; by contrast, in the molecular diseasome two diseases are linked if they share associated genes/proteins [13]. We hypothesised that, if comorbid diseases in COPD exacerbation patients share molecular pathways, many of the relationships seen at the clinical level (clinical diseasome) should be reproduced at the molecular level (molecular diseasome).
Methods
Study design, population and ethics
We used information collected from 5447 COPD patients hospitalised because an exacerbation of COPD included in two large clinical audits [14–17]. For this analysis, we used those comorbidities included in the Charlson Comorbidity Index (online table S1) [19], as per the information contained in the clinical records, which admittedly may be incomplete. To avoid spurious correlations, we investigated only those diseases whose prevalence in the study population was >1% [18]. The ethics committees of all participating institutions approved the study.
Clinical diseasome
As illustrated in figure 1a, in the clinical diseasome two diseases are linked if: 1) their frequency of co-occurrence is significantly higher than that expected by chance (p<0.05) assuming a Poisson distribution (online table S2); and 2) they show both a relative risk (RR) >1 and a Phi correlation coefficient (ϕ) >0 [18]. Cytoscape was used to represent the clinical diseasome graphically, in which node size is proportional to disease prevalence, node colour represents disease type (online table S3), and edge thickness is proportional to ϕ [20].

Graphical representation of the methodology used to construct the a) clinical diseasome and b) molecular diseasome. Further explanations can be found in the text.
Molecular diseasome
As shown in figure 1b, in the molecular diseasome two diseases are linked if they: 1) share disease-associated genes, as identified by DisGeNET, a database that integrates information on gene−disease associations from various public repositories and the biomedical literature [21, 22]; and/or 2) the proteins encoded by disease-associated genes are connected in the interactome [23], a publically available protein interaction network (HIPPIE) [24]. As detailed in the online supplement, to reduce the potential bias that shared genes/proteins might be more easily identified in those diseases that have been more extensively characterised, and to estimate the strength of the association between two diseases in the molecular diseasome, we used the Molecular Comorbidity Index (MCI) [13] and a bootstrap analysis. Cytoscape software was also used to represent the molecular diseasome graphically, in which node size is proportional to the number of associated genes, node colour represents disease type (online table S3), and edge thickness is proportional to the MCI [13].
Analysis of shared genes
As detailed in the online supplement, we calculated the percentage of genes shared by disease pairs that could be identified both in the molecular and clinical diseasomes. Besides, we used gene ontology enrichment analysis to explore the existence of common biological mechanisms in genes associated with individual diseases [25].
Statistical analysis
Results are presented as mean±sd, proportion or range, as appropriate. The statistical significance of differences between groups was assessed using one-way ANOVA, Mann−Whitney and Chi-square with Yates correction tests, as appropriate. p-values were corrected (Bonferroni) for multiple comparisons (n=11 per clinical diseasome) and were considered significant only if p<0.005.
Results
Participant characteristics
Table 1 presents the main characteristics of the 5447 COPD patients included in the analysis, stratified by the number of comorbidities present. There was a clear male preponderance and age and body mass index (BMI) increased slightly (but significantly) in those with more comorbidities (table 1). Airflow limitation was severe or very severe (Global Initiative for Chronic Obstructive Lung Disease (GOLD) grades 3–4) in the majority of COPD patients (81%). Interestingly, it was slightly but significantly worse in patients without concomitant disorders, who were also younger (table 1).
Demographic, clinical and functional characteristics of participants stratified by number of concomitant disorders present
Figure 2 shows the prevalence (figure 2a) and frequency distribution (figure 2b) of comorbidities present in the studied population of patients hospitalised owing to an exacerbation of COPD. Hemiplegia, AIDS, leukaemia and lymphoma were excluded from further analysis in order to avoid spurious correlations (their prevalence was <1% [18]; see Methods). Final analysis, therefore, included 11 out of the 14 diseases listed in the Charlson Comorbidity Index (table S1).

a) Prevalence and b) frequency distribution of the number of comorbidities in the patients studied. The red line indicates the threshold prevalence of 1% used to filter low-prevalence diseases for the analysis. Further explanations can be found in the text.
Clinical diseasome in exacerbation of COPD
A total of 55 disease pairs are combinatorially possible for the 11 diseases considered. We observed that the prevalence of 23 of these pairs (42%) was higher than expected by chance (table S2). These 23 disease pairs therefore constitute the clinical diseasome of the patients studied (figure 3a). Figure 3c (red line) shows that the frequency distribution of the node degree (k), this is the number of links of each node, was bimodal in the clinical diseasome, with a group of poorly connected diseases (n≤2; connective tissue disease, neoplasms, liver and dementia) and another group that was highly connected (n≥5; cardiovascular diseases, kidney, diabetes and ulcer). The mean±sd of k was 4.2±2.6.
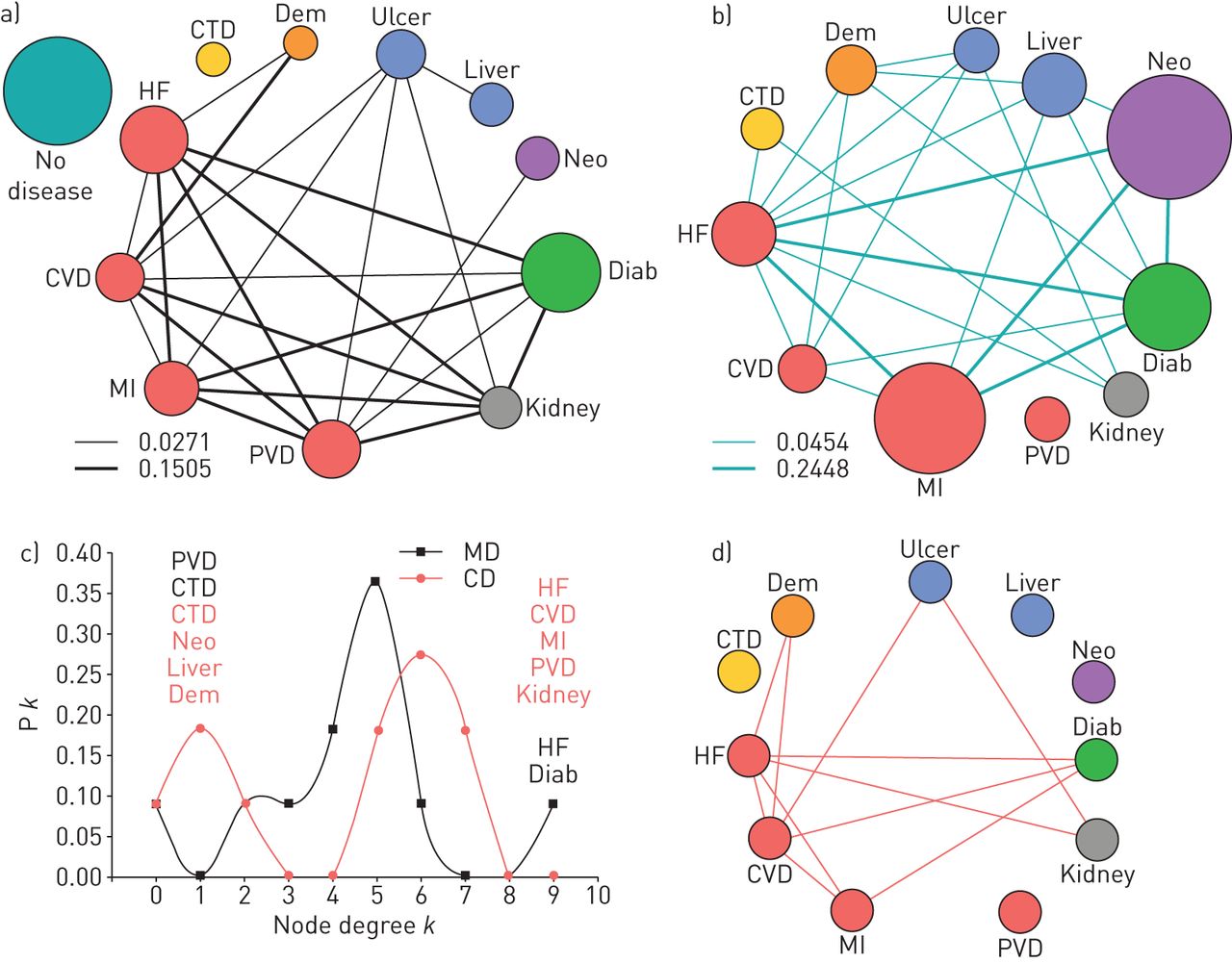
a) Clinical diseasome of patients with exacerbation of chronic obstructive pulmonary disease (COPD). Node colours indicate disease category (table S3), node size is proportional to disease prevalence and link width is proportional to the strength of correlation (ϕ). b) Molecular diseasome of patients with exacerbation of COPD. Node colours also indicate disease category, node size is proportional to number of associated genes/proteins, and link width is proportional to the Molecular Comorbidity Index value. c) Frequency distribution (Pk) of node degree (k) in the clinical diseasome (CD; red circles) and in the molecular diseasome (MD; black squares); disease names indicate if they correspond to the clinical (red) or the molecular diseasome (black). d) Shared links between the clinical and molecular diseasome in the patients studied. Further explanations can be found in the text.
Molecular diseasome
We found that 25 of the 55 potentially maximal disease pairs (46%) were included in the molecular diseasome after the bootstrap analysis of MCI values (figure 3b). The number of associated genes/proteins per disease (as indicated by the node diameters in figure 3b) ranged from less than 15 (connective tissue disease, n=5; ulcer, n=11; kidney diseases, n=12) to more than 200 (cancer, n=288; myocardial infarction, n=242), with a mean±sd value of 92±104. Figure 3c (black line) presents the frequency distribution of the node degree (k) in the molecular diseasome, which shows that not all diseases were equally interconnected, ranging from peripheral vascular disease that appears isolated (k=0) to heart failure, which was highly connected (k=9). The mean±sd of the k values was 4.5±2.2.
Clinical−molecular diseasome comparison and gene ontology enrichment analysis
As shown in figure 3d the clinical and molecular diseasomes shared 11 links. This means that 11 out of 23 (47.8%) comorbidity pairs in patients with COPD exacerbation can be explained by shared genes/proteins. A total of 89 genes were associated with these 11 disease pairs, 22 of which (24.7%) were identified in more than one disease pair (table 2). The genes of IL-1β (interleukin-1β), EDN1 (endothelin 1) and ACE (angiotensin converting enzyme-1) were often shared by comorbidity pairs. Further, the 11 disease pairs shared by the molecular and clinical diseasomes in exacerbation of COPD (figure 3d) included seven distinct diseases. The ontology enrichment analysis of the genes associated with each of these seven diseases showed that six of them (not ulcer) shared 12 gene ontology biological terms (figure S1).
List of genes that support a common molecular basis between comorbidity pairs observed in patients with exacerbation of chronic obstructive pulmonary disease
Influence of age, smoking and airflow limitation severity
To explore the influence of age on the clinical diseasome of the studied patients, we compared the clinical diseasome of patients included in the lowest (<68 years; n=1706) and highest (>77 years; n=1779) age tertiles. We observed that the prevalence of all comorbid diseases (i.e. node size) was significantly higher in older patients (figure S2a and S2b; table S4) except that of liver disease, which was higher in younger patients. The remaining quantitative attributes of both clinical diseasomes were similar (table S5), as was the k distribution (figure S2c). Only one link (neoplasm−peripheral vascular disease) was differentially present in the clinical diseasomes of relatively younger and older patients with exacerbation of COPD (figure S2d).
Figure S3 compares the clinical diseasome of COPD patients with ≤20 pack-year (n=356) and >50 pack-year (n=1743) cumulative smoking exposure. These thresholds were set arbitrarily in order to compare groups of patients with relatively low and high smoking exposure (lower thresholds resulted in a very small group of subjects). As shown in table S5, the differences between the mean node diameters (proportional to prevalence of diseases) were not statistically significant between the two groups (10.5±6.5 versus 10.5±6.1), neither was the node degree k (2.9±2.1 versus 4.2±2.6). However, the clinical diseasome of COPD patients with lower smoking exposure had a lower number of links (16 versus 23). This was reflected in a rightward shift of the k distribution curve in patients with high smoking exposure (figure S3c). Likewise, figure S3d shows seven differential links between the two groups of patients, which was the highest number observed in the comparisons performed in this study (figures S2d and S3d).
Finally, the clinical diseasome of patients with different airflow limitation severity (GOLD grade 1–2 (n=1051) versus GOLD grade 3–4 (n=4396)) was remarkably similar (figure 4). The total number of connected nodes in the network, average node degree (k), and strength of the association of diseases (ϕ) were similar in both groups of patients (table S5). Likewise, the k frequency distribution was bimodal in both groups (figure 4c). The only notable exception was that GOLD grade 3–4 patients had two additional disease pairs in their clinical diseasome (ulcer−cardiovascular disease and ulcer−myocardial infarction) (figure 4d), and that neoplasms were also more prevalent in these patients (2% versus 7%; p<0.0001; odds ratio 0.269) (table S4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical diseasome of chronic obstructive pulmonary disease (COPD) patients with a) Global Initiative for Obstructive Lung Disease (GOLD) grade 1–2 and b) GOLD grade 3–4. Asterisks indicate differences in prevalence (Chi-square; p<0.005). The node labelled “no disease” indicates, for comparison, the prevalence (i.e. diameter) of COPD patients without concomitant diseases. The width of the links is proportional to co-occurrence prevalence (two examples are provided at the bottom of the panel). c) Frequency distribution (Pk) of node degree (k) in GOLD grade 1–2 (black squares) and GOLD grade 3–4 (red circles). Specific diseases with low and high k values are indicated. d) Differential disease links in the clinical diseasome of GOLD 3–4 versus GOLD 1–2 patients. Further explanations can be found in the text.
Discussion
This paper uses network analysis [6, 7, 13, 18] to explore the molecular basis of comorbidities in patients hospitalised owing to an exacerbation of COPD. Our results provide three major novel findings: 1) by comparing their clinical and molecular diseasomes, we observed that about half of the disease pairs identified in their clinical diseasome (47.8%) could also be identified in the molecular diseasome, suggesting shared molecular mechanisms across comorbidities; 2) we identified genes related to inflammation and vascular tone regulation (table 2), and biological processes related to the regulation of homeostasis, response to oxygen containing compounds and lipid metabolism (figure S1), as those most commonly shared; and, finally, 3) we observed that the clinical diseasome of these patients is, by and large, independent of age (within the age range studied here), cumulative smoking exposure or severity of airflow limitation.
Previous studies
It is well established that comorbidities occur often in COPD patients and impact their clinical course negatively [1–5, 26]. Divo et al. [27] recently proposed the term comorbidome for the combined graphic representation of their prevalence and associated risk of death. Vanfleteren et al. [3] used self-organising maps to identify five clusters of 13 objectively identified comorbidities in COPD patients included in a rehabilitation programme. Very recently, Divo et al. [11] used network analysis to explore the clinical diseasome of 1969 clinically stable COPD patients. Neither of these two studies, however, investigated the potential molecular basis of these comorbidities [13]. However, our group has recently applied a bioinformatic analysis on publicly available data to explore the molecular relationships between COPD and frequent comorbid diseases [13]. The study reported here extends these previous studies by investigating, first, the biological basis of COPD comorbidities themselves using a real-life, convenient, large, clinical audit database of hospitalised patients with COPD exacerbation and, secondly, the potential influence of age, cumulative smoking exposure and severity of airflow limitation.
Interpretation of findings
The observation that about half of the disease pairs identified in the clinical diseasome were also identified in the molecular diseasome suggests that shared molecular mechanisms (figure S1) are likely to be pathogenically relevant [8]. Interleukin-1β, angiotensin converting enzyme-1 and endothelin-1 genes were associated with more than 45% of these shared links (table 2), supporting the role of inflammation and vascular physiology as specific mechanisms underlying the pathobiology of comorbidities in COPD. Furthermore, these three genes have also been related with the pathogenesis of COPD [28–34], hence providing a link between comorbidities and COPD itself, as recently suggested by a model of COPD that identifies the lung, the bone marrow and the adipose tissue as a vascularly interconnected network of potential pathogenic relevance [35]. In this context, it is of note that the diseasome (clinical or molecular diseasome) presented here has a very prominent “vascular” component, which may reflect the fact that admissions for exacerbation of COPD can also have a significant cardiovascular component [36, 37].
Conversely, however, the other half of disease pairs identified in the clinical diseasome were not mirrored in the molecular diseasome, and vice versa (figure 3). Several explanations may be conceived in this case. First, current knowledge about the molecular mechanisms of the considered diseases is likely to be incomplete [38]. Secondly, molecular mechanisms not considered in our analysis, such as miRNA regulation and other epigenetic changes, gene expression changes and/or indirect regulations through metabolic networks and other signalling pathways [39] may contribute to explain this. Thirdly, shared non-molecular risk factors (e.g. environmental exposures) can contribute to disease associations [13, 40]. For instance, disease associations can also be (partially) the result of undesired therapeutic side-effects [41]. Finally, it is also possible that some gene variants have insufficient penetrance to cause clinical illness in the studied patients.
Finally, we failed to identify a significant effect of age and cumulative smoking exposure on the clinical diseasome of these patients with exacerbation of COPD (figures S2 and S3, respectively). The relatively narrow age range of the studied population, as well as the fact that all participants had been substantially exposed to smoking (table 1), might have limited our ability to identify any potential effect on their clinical diseasome. On the other hand, the observation that the clinical diseasome is largely independent of the GOLD grade of airflow limitation (figure 4) is in keeping with previous studies showing that prevalence of comorbid diseases in COPD is similar in patients with different degrees of airflow limitation [5, 26], and supports the concept that lung abnormalities in COPD are not likely driving the occurrence of multimorbidity [8] and that we should move towards a “Copernican view” of COPD, where the lung disease is only one of a number of age-related prevalent diseases [7, 42–44]. The observation (figure 4) that the comorbid diseases studied here share many biological processes with COPD further reinforces this hypothesis.
Strengths and limitations
The use of network analysis [6, 7, 11, 45] to investigate the molecular basis of comorbidity in COPD represents a step forward in our understanding of this complex disease [46, 47] and a strength of this paper. Yet, we acknowledge that it has a number of limitations that deserve comment. First, the audit nature of the two convenient and large case series of patients hospitalised because of COPD exacerbation used for analysis [14–17] did not allow the active search of comorbidities [3], so we had to identify them by careful review of clinical records. This might contribute to explaining why 38% of the patients studied here did not apparently suffer any comorbidity, in contrast with previous studies [5, 27, 48]. Likewise, potentially relevant comorbidities not listed in the Charlson Comorbidity Index were not included in the analysis, nor was clinical severity. Secondly, our patients were recruited during an episode of hospitalisation because of ECOPD exacerbation, so their clinical diseasome might be different under conditions of clinical stability. Yet, given that comorbidities develop chronically, we believe that our results reflect the clinical diseasome of COPD at large and are unrelated to the exacerbation episode itself. Thirdly, most COPD patients were males (table 1). Given that the inflammatory response to smoking appears different in males and females [49], whether the clinical diseasome described here in males with COPD is different in female patients deserves further investigation. Similarly, the relatively narrow spectrum of patients and the limited number of comorbidities included in this analysis limit the generalisability of our findings [43, 44]. Finally, while the use of publicly available databases is efficient and economical [50], the molecular and clinical links may change depending on the genetic background of patients and their environment. Because the current cohort originates from a clinical audit, it is not suitable for in vivo direct molecular measurements, which are required for the validation of our observations.
Conclusions
About half of the disease pairs identified clinically in patients hospitalised because of an exacerbation of COPD share molecular mechanisms, in particular those related to inflammation and vascular tone regulation, that can explain their coexistence. The clinical diseasome of these patients appears to be basically independent of age, cumulative smoking exposure and/or airflow limitation severity.
Acknowledgements
Authors thank all field investigators of the AUDIPOC study as well as those involved in the Spanish branch of the European COPD audit (see Appendix to the online supplement).
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: The study was supported in part by Instituto de Salud Carlos III-Fondo Europeo de Desarollo Regional (grants CP10/00524, PI13/00082, PI09/00629, PI12/01117), the Innovative Medicines Initiative Joint Undertaking under grant agreements 115191 (Open PHACTS) and 115372 (EMIF), resources of which are composed of financial contribution from the European Union's Seventh Framework Program (FP7/2007-2013) and EFPIA companies' in kind contribution. FUCAP-2013, SEPAR PI192/2012 and PI065/2013 also supported this study. Funding information for this article has been deposited with FundRef.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received March 24, 2015.
- Accepted June 11, 2015.
- Copyright ©ERS 2015