



Efficacy and safety of inhaled ENaC inhibitor BI 1265162 in patients with cystic fibrosis: BALANCE-CF 1, a randomised, phase II study
- Christopher H. Goss1⇑,
- Isabelle Fajac2,
- Raksha Jain3,
- Wolfgang Seibold4,
- Abhya Gupta4,
- Ming-Chi Hsu5,6,
- Sivagurunathan Sutharsan7,
- Jane C. Davies8,9 and
- Marcus A. Mall10,11,12
- 1Dept of Medicine, Dept of Pediatrics, University of Washington, Seattle Children's Hospital and Research Institute, Seattle, WA, USA
- 2AP-HP, Université de Paris, Paris, France
- 3Dept of Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA
- 4Boehringer Ingelheim, Biberach, Germany
- 5Boehringer Ingelheim, Shanghai, China
- 6Shanghai Junshi Biosciences Co. Ltd, Shanghai, China
- 7Division for Cystic Fibrosis, Dept of Pulmonary Medicine, University Medicine Essen – Ruhrlandklinik, Essen, Germany
- 8National Heart and Lung Institute, Imperial College London, London, UK
- 9Paediatric Respiratory Medicine, Royal Brompton and Harefield Hospitals, London, UK
- 10Dept of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine, Charité – Universitätsmedizin Berlin, Berlin, Germany
- 11Berlin Institute of Health (BIH), Berlin, Germany
- 12German Center for Lung Research (DZL), associated partner site, Berlin, Germany
- Corresponding author: Christopher Goss (CGoss{at}medicine.washington.edu)
Abstract
Background Inhibition of the epithelial sodium channel (ENaC) in cystic fibrosis (CF) airways provides a mutation-agnostic approach that could improve mucociliary clearance in all CF patients. BI 1265162 is an ENaC inhibitor with demonstrated pre-clinical efficacy and safety already demonstrated in humans.
Objective We present results from BALANCE-CFTM 1, a phase II, placebo-controlled, randomised, double-blind study of four dose levels of BI 1265162 versus placebo for 4 weeks on top of standard of care in adults and adolescents with CF.
Results Initially, 28 randomised subjects (BI 1265162 200 µg twice daily n=14, placebo twice daily n=14) were assessed at an interim futility analysis. Compared with placebo, numerical changes of –0.8% (95% CI –6.6 to 4.9%) in percentage predicted forced expiratory volume in 1s (ppFEV1) and +2.1 units (95% CI –2.4 to 6.5 units) in lung clearance index (LCI) were observed in the active group, meeting a pre-defined stopping rule; accordingly, the study was terminated. Recruitment had continued during the interim analysis and pending results; 24 patients were added across three dose levels and placebo. The final results including these patients (+1.5% ppFEV1, 200 µg twice-daily dose versus placebo) were not supportive of relevant clinical effect. Furthermore, LCI change was not supportive, although interpretation was limited due to insufficient traces meeting quality criteria. A 9.4-point improvement in the Cystic Fibrosis Questionnaire – Revised Respiratory Domain was observed in the 200 µg twice daily dose group versus placebo. BI 1265162 up to 200 µg twice daily was safe and well-tolerated. Pharmacokinetics were similar to those in healthy volunteers.
Conclusion BI 1265162 was safe, but did not demonstrate a potential for clinical benefit. Development has been terminated.
Abstract
Phase I trials showed that single and multiple doses of the inhaled ENaC inhibitor BI 1265162 are safe. In this phase II trial in patients with CF, BI 1265162 was safe, but did not demonstrate clinically relevant efficacy. The trial was terminated. https://bit.ly/3CiB8uM
Introduction
Cystic fibrosis (CF) is a multisystem, life-threatening, autosomal recessive genetic disease resulting from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which encodes the apical cell membrane CFTR anion channel protein [1, 2]. Mutations in CFTR result in a defective or absent ion channel that secretes reduced levels of chloride and bicarbonate [1–4]. CFTR dysfunction and/or proteolytic activation by host- and bacteria-derived proteases in CF lead to hyperactivation of the epithelial sodium channel (ENaC) [5–11]. In turn, this leads to reduced airway surface liquid volume, dehydrated mucus and dysfunctional cilia, resulting in poor mucociliary clearance (MCC) [1, 12]. Poor MCC leads to mucus obstruction, chronic airway inflammation and infection with bacterial pathogens [13].
CFTR modulators address the underlying ion transport defect in CF [14]. Currently, approved CFTR modulators include the potentiator ivacaftor (for patients with at least one G551D allele, other CFTR gating mutations and responsive mutations based on clinical and/or in vitro assay data); the corrector/potentiator combinations lumacaftor/ivacaftor (for patients homozygous for the F508del mutation) and tezacaftor/ivacaftor (for patients homozygous for the F508del allele, those with an F508del allele plus residual-function mutation and responsive mutations based on clinical and/or in vitro assay data); and the triple-agent CFTR modulator elexacaftor/tezacaftor/ivacaftor (for patients with at least one F508del allele and responsive mutations based on in vitro assay data). In clinical studies, CFTR modulators have improved percentage predicted forced expiratory volume in 1 s (ppFEV1) by 3–14% [15–22], with a sustained effect confirmed in open-label extension studies [23, 24]. A real-world study has demonstrated a slowed decline of ppFEV1 over 5 years [25].
However, for most patients with CF, an improvement in pulmonary function is not necessarily a return to normal, and exacerbations still occur, albeit at a lower rate [15, 24, 25]. In addition, bacteria are not eradicated from the airways over time [25–27]. Treatments that target ENaC in addition to CFTR modulators could assist in further normalising airway surface hydration [28] by providing an enhanced electrical driving force favouring CFTR-mediated chloride secretion, restoring ion and water homeostasis [1, 29]. Furthermore, CFTR modulator therapy is not approved for ∼5–10% of patients with CF, because their mutations lead to an unresponsive CFTR protein [30]. In countries such as Brazil, Israel, Italy and Turkey, >30% of patients with CF do not possess an F508del allele [31, 32]; ENaC inhibition in these regions represents an even more significant therapeutic option. Therefore, ENaC inhibition is an important, mutation-agnostic therapeutic approach that could operate independently of CFTR function and mutation class [1, 30].
BI 1265162 is an ENaC inhibitor inhaled via the Respimat® Soft MistTM inhaler (SMI). BI 1265162 has demonstrated pre-clinical efficacy [33] and safety in healthy volunteers [34]. The objectives of this study were to assess the efficacy, safety and pharmacokinetics of 20 µg, 50 µg, 100 µg and 200 µg twice-daily doses of BI 1265162 (BI 20, BI 50, BI 100 and BI 200, respectively) via the Respimat SMI, compared with placebo twice daily (PBO), as an add-on to standard CF therapies in patients aged ≥12 years.
Methods
A summary of methods is provided here. A full description can be found in the supplementary material.
This was a multicentre, multinational, randomised, double-blind, placebo-controlled, parallel-group, dose-ranging study (figure 1). 98 patients aged ≥12 years were planned for randomisation. The start of adolescent patients' enrolment was to be based on review of adult safety data, carried out by an independent data monitoring committee in collaboration with the CF Foundation.

Study design. R: randomisation; CF: cystic fibrosis.
The primary end-point was the change from baseline after 4 weeks of treatment in trough (30 min pre-dosing) ppFEV1. Secondary end-points were change from baseline after 4 weeks of treatment in: 1) lung clearance index (LCI); 2) Cystic Fibrosis Questionnaire – Revised (CFQ-R) [35] total score; and 3) Cough and Sputum Assessment Questionnaire (CASA-Q) [36], adverse events and pharmacokinetics.
An interim futility analysis on the first 28 patients (BI 200 or PBO) was planned to assess potential for efficacy and to prevent exposure of further patients in case of insufficient potential. Per protocol, recruitment continued pending results of the interim analysis to enable the study to be carried out in the most time-efficient manner. A decision on termination was to be made if the increase in trough ppFEV1 was <1.5% and the decrease (improvement) in LCI was <0.3 units (futility).
The planned analyses for proof of concept and dose finding were to use multiple comparison and modelling techniques to measure the difference between PBO and active treatment. Power calculations for the final analysis were to be based on having ppFEV1 results for ≥24 evaluable patients each for the BI 200 and PBO groups and ≥12 evaluable patients each for all other groups.
A restricted maximum likelihood-based approach using a mixed model with repeated measurements (MMRM) was carried out to assess the change from baseline in trough ppFEV1. Visits were treated as the repeated measure with an unstructured covariance structure used to model the within-patient measurements. Analysis of covariance (ANCOVA) with adjustment for categorical effects of treatment and the fixed continuous effect of baseline was carried out to assess change from baseline in LCI. Patient-reported outcomes were descriptive in nature.
Pre-specified sensitivity analyses to address any outlier data points and expected variability were carried out for both ppFEV1 and LCI end-points. Data were reviewed by an interim analysis assessment committee (Boehringer Ingelheim internal, independent from the study team) at the interim futility analysis for the impact of outliers.
A model-based pre-defined subgroup analysis was performed to investigate any impact of patient characteristics, CFTR mutation status and concomitant CF therapy use on the change from baseline in trough ppFEV1.
Results
Study population
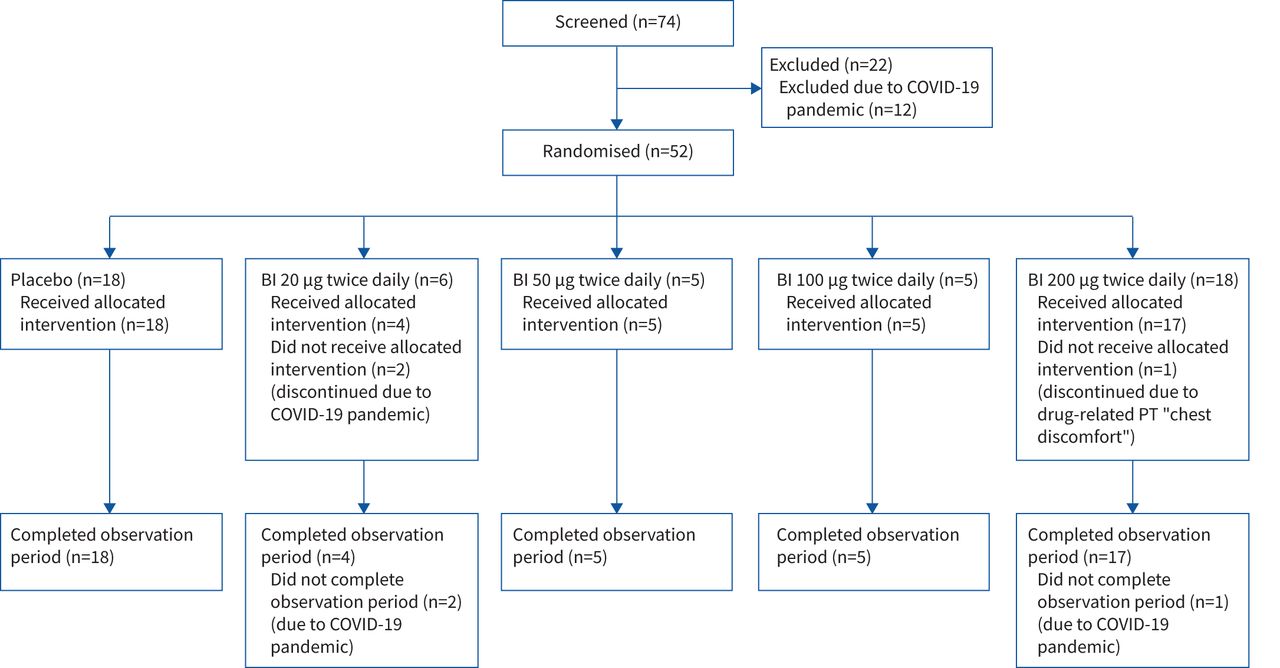
Patient disposition is described in figure 2. Baseline characteristics and medication use were balanced between groups and are summarised in table 1.
{kind=link}
{kind=link}
Patient disposition. COVID-19: coronavirus disease 2019; BI: BI 1265162; PT: preferred term.
Patient baseline demographics and concomitant drug use: treated set
Due to the coronavirus disease 2019 (COVID-19) pandemic, there was a temporary halt in recruitment just prior to the interim futility results. This further added to the limitation in sample size beyond the interim analysis. A total of 52 patients were randomised into the PBO and BI 20, 50, 100 and 200 dosing groups (n=18, n=6, n=5, n=5 and n=18, respectively) until termination. 49 (94.2%) patients completed the planned treatment and observation periods. Three (5.8%; two receiving BI 20 and one receiving BI 200) prematurely discontinued study medication and did not complete the planned observation period, due to the COVID-19 pandemic (BI 20) and adverse events (BI 200). Embryo–foetal development data were not available at study start, so that women of childbearing potential (WoCBP) were excluded in the initial protocol, leading to a male-predominant population; embryo–foetal development data allowed inclusion of WoCBP using adequate contraception in a revision of the protocol (supplementary material). Treatment compliance was high, with mean±sd percentages of prescribed medication taken during the treatment period ranging from 93.2±17.7% in the BI 20 group to 100.4±4.8% in the BI 100 group, with no relevant difference between groups.
All enrolled patients were adults. Enrolment of adolescents was approved by the independent data monitoring committee, but was not possible because of the recruitment stop due to the COVID-19 pandemic and the interim futility analysis.
Efficacy
All efficacy data presented below are after 4 weeks’ treatment.
Interim analysis
Results from the interim analysis of BI 200 versus PBO for ppFEV1 and LCI (n=14 versus n=14, and n=3 versus n=6, respectively) are presented in table 2.
Change in trough percentage predicted forced expiratory volume in 1 s (ppFEV1) after 4 weeks of treatment with BI 1265162 (BI) 200 µg twice daily: interim analysis
An adjusted mean±se decrease in trough ppFEV1 of 0.1±1.95% was observed in the BI 200 group compared with a 0.7±2.00% increase in the PBO group, equating to a numerical difference of –0.8% (95% CI –6.6 to 4.9%).
An adjusted mean±se increase in LCI of 0.8±1.46 units was observed in the BI 200 group compared with a decrease of 1.3±1.01 units in the PBO group, equating to a numerical difference of 2.1 (95% CI –2.4 to 6.5) units.
Stopping rules defined for the futility analysis were met for this study; recruitment was stopped and the study terminated when these data were available, which was concurrent to when recruitment had already been placed on hold due to the COVID-19 pandemic. Thus, hypothesis testing was not carried out and sample size was not adequate to assess the dose–response relationship. Statistical analysis of ppFEV1 and LCI is exploratory and descriptive only, and inferences should be made with caution.
Final analysis
Results from the final analysis of treatment with BI 200 (n=16) versus PBO (n=18) for ppFEV1, including sensitivity analyses, are presented in table 3. At study baseline, mean±se ppFEV1 was 59.21±2.09%. An adjusted mean±sd increase in trough ppFEV1 of 0.5±1.77% was observed in the BI 200 group compared with –1.0±1.70% in the PBO group, equating to a numerical difference of 1.5% (95% CI –3.5 to 6.5%).
Change in trough percentage predicted forced expiratory volume in 1 s (ppFEV1) and lung clearance index (LCI) after 4 weeks of treatment with BI 1265162 (BI) 200 µg twice daily: final analysis
Descriptive and exploratory statistics for change in trough ppFEV1 for all groups are shown in table 4 and supplementary figure S1a. A numerical mean increase from baseline in trough ppFEV1 was observed in the BI 100 and 200 groups. Trough ppFEV1 was relatively unstable in the PBO and BI 200 groups over the 4-week period (variability extremes of +17.4% and –15.2%, and +11.4% and –12.7% in lung function changes, respectively). Individual patient changes from baseline in ppFEV1 are shown in supplementary figure S2.
Change in trough percentage predicted forced expiratory volume in 1 s (ppFEV1) and lung clearance index (LCI) after 4 weeks of treatment with BI 1265162 (BI) twice daily: all treatment groups (descriptive statistics)
In a sensitivity analysis, five and four patients from the BI 200 and PBO groups, respectively, had ppFEV1 visit data censored due to adverse events that could have affected lung function, unacceptable pulmonary function test quality or poor treatment compliance (supplementary table S1). The decision to censor the data was made without knowing treatment allocation. Sensitivity analyses did not change the outcome of either the interim or final analyses (a numerical difference in ppFEV1 between the BI 200 and PBO groups of 2.7% (95% CI –2.3 to 7.7) in the MMRM analysis and 5.7% (95% CI –1.6 to 12.9) in the quantile regression analysis). Individual patient changes from baseline in ppFEV1 in the sensitivity analyses are shown in supplementary figure S3.
Subgroup analysis showed a consistent response pattern of trough ppFEV1 after treatment with BI 1265162 across all subgroups, but no responsive subpopulations were identified (supplementary figure S4). A total of 19 (36.5%) out of 52 patients were receiving CFTR modulator therapy at randomisation (seven (38.9%), three (50.0%), two (40.0%) and seven (38.9%) patients in the placebo, BI 20, BI 50 and BI 200 groups, respectively). In the subgroup analysis, patients on BI 200 receiving CFTR modulators demonstrated a mean numerical –1.2% (95% CI –8.8% to 6.4%) change in ppFEV1 compared with placebo, whereas patients not receiving CFTR modulators in this group demonstrated a numerical 3.1% (95% CI –4.2% to 10.3%) change in ppFEV1 compared with placebo (supplementary figure S4). The confidence intervals of the subgroups were overlapping.
At study baseline, 16 patients performed valid LCI tests, with a mean±se score of 14.68±1.06 units. At week 4, only 11 patients performed valid LCI tests; treatment with BI 200 (n=3) resulted in an adjusted mean±se increase in LCI of 0.8±1.46 units, compared with a decrease of 1.3±1.01 units in the PBO group (n=6; ANCOVA analysis), equating to a numerical difference of 2.1 units (95% CI –2.4 to 6.5).
Descriptive and exploratory statistics for change in LCI for all groups are shown in table 4 and supplementary figure S1b. Supplementary figure S5 describes individual patient changes from baseline in LCI. The LCI analysis is limited given the small number of LCI values that could be obtained across the study.
Patient-reported outcomes
The mean CFQ-R total score increased (improved) for all groups except BI 100 (supplementary table S2). For CFQ-R Respiratory Domain, the BI 20, 100 and 200 groups met the minimal clinically important difference (+4 points) outcomes for patients with stable CF [37] (mean±sd scores 6.94±5.32, 6.67±13.26 and 6.60±14.93, respectively).
There was no correlation between change in CFQ-R Respiratory Domain score and change in ppFEV1 (supplementary figure S6), but sample sizes were limited.
The mean Cough and Sputum Symptom Domain score of the CASA-Q increased, showing numerical improvement for patients across all groups; however, no consistent dose-dependent trends were observed with no apparent dose dependence (supplementary table S3).
Safety
Overall adverse events are summarised in table 5. Drug-related adverse events were reported for 16.7%, 0%, 20.0%, 20.0% and 27.8% of patients in the PBO and BI 20, 50, 100 and 200 groups, respectively. There was a low incidence of CF exacerbations (one (5.6%) out of 18 patients in each of the placebo and BI 200 groups).
Overall summary of patients with adverse events (treated set)
Adverse events for more than one patient in any treatment group are detailed in table 6. An adverse event of special interest (AESI), hyperkalaemia, was reported for two patients (PBO n=1; BI 200 n=1). This was not considered serious and did not lead to dose reduction or discontinuation. One patient in the BI 200 group discontinued due to chest discomfort of mild intensity on study days 2–4. This was considered to be drug related by the investigator. However, this event was not considered a serious adverse event (SAE) or an AESI. Two patients had SAEs (BI 200 n=1 (lung congestion); PBO n=1 (hypoglycaemia with a fatal outcome after the end of the treatment period)).
Adverse events (preferred terms) reported for one or more patients in any treatment group (treated set)
Pharmacokinetics
Results of pharmacokinetics analyses are shown in supplementary table S4. Steady-state mean concentration profiles at day 8 (visit 3) showed fast absorption across all groups. Mean maximal concentration (Cmax) and area under the concentration–time curve from 0 to 4 h (AUC0–4) at visit 3 increased almost proportionally for the BI 20, 50 and 100 groups. The mean trough concentrations of BI 1265162, as well as drug concentrations at 5 min after inhalation (C0.083), were similar across individual patients and groups, with some exceptions. The variability for Cmax and AUC0–4 was high for the BI 200 group (81.5% and 71.0% geometric coefficient of variance, respectively), and lower for the other groups (ranging from 20.1% and 8.93%, respectively, in the BI 100 group to 57.0% and 45.3%, respectively, in the BI 20 group).
Discussion
The aim of the study was to investigate the efficacy, safety and pharmacokinetics of the ENaC inhibitor BI 1265162 in adult and adolescent patients with CF versus placebo.
The independent data monitoring committee proposed to enrol adolescents, but due to a COVID-19 pandemic-driven stop of enrolment and then termination of the study based on results of a futility analysis, adolescent patients were not enrolled. In addition, due to the early stopping of the study, sample sizes, especially in the lower-dose groups, were small, and no hypothesis testing of dose–response could be carried out.
Due to an insufficient effect on trough ppFEV1 and LCI after 4 weeks of treatment at an interim futility analysis, and limited potential for effect in the sensitivity analyses, the study was terminated. In addition, there was no significant effect in the larger dataset of completed patients (including those enrolled during the analysis of interim data). No response characteristics could be identified. Subgroup analysis in this study did not suggest an impact of concomitant stable CFTR modulator therapy on ppFEV1 changes seen with treatment with BI 1265162. However, small sample sizes of the subgroups do not allow any stringent conclusion. No dose-dependent trends in improvements in patient-reported outcomes were observed, although clinically relevant changes compared with PBO were observed for the BI 20, 100 and 200 groups. There was no correlation between change in ppFEV1 and change in CFQ-R Respiratory Domain scores at 4 weeks, although the sample size was relatively small. Improvement in patient-reported outcomes is not always correlated with improvements in lung function. In a phase Ib study of the antisense oligonucleotide eluforsen in patients with F508del/F508del CF, at least minimal clinically important difference (+4 points) in CFQ-R Respiratory Symptom score was achieved in two dose groups of a multiple-ascending-dose cohort compared with placebo, but this was not related to any meaningful change in ppFEV1 [38]. In an analysis of lung function changes and signs and symptoms of pulmonary exacerbations in patients with CF in the Standardized Treatment of Pulmonary Exacerbations study, only an extremely weak correlation between ppFEV1 and Chronic Respiratory Infection Symptom Score (R2=0.157; p<0.001) was observed [39].
Occurrence of drug-related adverse events was similar, and occurrence of CF exacerbations was low, across treatment groups. No clinically relevant changes from baseline in vital signs and physical examinations were observed. Occurrence of drug-related adverse events was low and comparable across PBO and BI 1265162 groups. As might be expected for patients with CF, the most frequently reported system organ classes were respiratory, thoracic and mediastinal disorders, and infections and infestations, which are commonly reported in studies of CF therapies and may be related to underlying disease. Two cases of hyperkalaemia were reported (PBO n=1; BI 200 n=1). This adverse event deserves special attention as it could be caused by renal activity of BI 1265162 due to high levels of ENaC expression in the kidney [1], and previous clinical development of ENaC inhibitors has been hampered by hyperkalaemia [29]. One patient in the phase I study of BI 1265162 had hyperkalaemia [34]; however, renal blockade of ENaC was considered unlikely given the urinary electrolyte values in that subject. The cases of hyperkalaemia reported in this study were not considered serious, and did not lead to dose reduction or discontinuation. The overall adverse event evaluation did not indicate a higher risk for respiratory or infectious adverse events in the active treatment arms.
On one hand, the ppFEV1 and LCI cut-off values at the interim analysis were based on statistical calculations of having a high probability for the study succeeding and achieving a clinically meaningful improvement, with n=14 each in the PBO and BI 200 groups based on the assumed treatment effect. On the other hand, the cut-off was chosen to have good chances to stop the trial early assuming no treatment effect. Based on the ppFEV1 signal observed at the interim, reaching a substantial lung function improvement was not expected to occur in this study with continued recruitment. The probability of achieving the original goal was re-evaluated conditioned on the observed results and number of patients (original analysis and including the additional patients) and confirmed a low probability of success even with the original assumptions for the treatment effect. A 9% predicted probability of reaching the targeted 4% improvement in ppFEV1 was calculated based on the available 52 randomised patients if the study had continued and fully recruited.
Previous failures of inhaled ENaC inhibitors in clinical studies may have been due to inadequate dosing and/or bronchiolar deposition in patients with heterogeneous airway plugging. The dose used in the current clinical study was based on fluid absorption data from a rat model (BI 1265162 was tracheally instilled) and MCC data from a sheep model (BI 1265162 was nebulised) [33], also correcting for lung deposition using the Respimat SMI in humans [40]. Nevertheless, underdosing in this study cannot be ruled out, without a more direct measure of ENaC function in the airways and because animal studies were carried out in models that had no mucus plugging or structural lung damage as seen in patients with CF. Therefore, the dose and duration of inhaled ENaC inhibitor required for a therapeutic benefit may have been underestimated.
This study had a number of adaptive steps that allowed early termination, with a number of design elements that could be considered or reconsidered for other studies.
Recruitment was continued during analysis of interim data. There must be a balance between expediting study completion with a potentially medically valuable drug and continued enrolment into a study of a non-efficacious drug. If efficacy had been greater, several months would have been saved in the programme; however, recruitment of almost half the study population into a study of a likely non-efficacious treatment regimen was avoided.
The decision to terminate was based on statistical considerations, which must be robust enough to handle individual variability, especially in small sample sizes. In our study, the standard deviations for ppFEV1 were as expected, and although a change from a Δ of –0.8% to 1.5% ppFEV1 was observed in the final analysis, the decision to stop the study after the futility analysis was considered correct given the very low probability of reaching the target ppFEV1 with the given study design (duration, dose, potential for efficacy).
As stated earlier, although overall variability was as expected, lung function in the placebo and BI 200 groups was unstable during the study, as indicated by the largest extremes in ppFEV1 values at week 4 of any treatment group. To increase lung function stability in future studies with potential for better treatment discrimination, an inclusion criterion of variability of ppFEV1 between screening and baseline of <15% could be considered. A longer stability period during run-in, for use of concomitant CF drugs, could also be considered.
A longer treatment period would leverage the usage of the MMRM approach and reduce the impact of missing data points, and also account for effects of temporary worsening that can occur in such a fluid disease.
The analysis of change from baseline in LCI contained data from only 20% of patients. This was due to eligibility criteria for this measurement (FEV1 >60% predicted) and quality-control requirements, which had been set and monitored in close collaboration with central over-reading centres (CORCs) to achieve the highest LCI quality. Of 28 patients who qualified for the N2 multiple-breath washout test at baseline, only 11 patients passed the quality-control test for LCI at both baseline and week 4 from a study population of 52. A number of measures could be implemented to further optimise LCI. Firstly, testing at screening (and not just baseline) would have provided: 1) a training opportunity for participants new to the technique; 2) rapid review of trace quality by the CORC to allow feedback to sites requiring technical improvements ahead of baseline visit; and 3) where LCI is a key outcome and protocol-defined, potential to use screening values in cases where the baseline visit test fails quality control. Secondly, in this study, LCI was performed at two visits (baseline and week 4). Having more than one “on-treatment” value would minimise any effect of missing data. Thirdly, operational challenges were experienced at some sites with less experience in carrying out the LCI test. When sample sizes are limited based on subgroup eligibility criteria, selecting the most highly skilled sites to perform this measurement would improve the proportion of successful attempts. Highly skilled sites are those that have consistently high success rates, know how to create a suitable testing environment, and observe any abnormalities and act on them accordingly. Finally, data from CFTR modulator studies have shown that LCI has superior sensitivity over FEV1 in early structural lung abnormalities associated with CF, particularly in younger patients [41–44]. In future studies, the utility of LCI will be better in mild-to-moderate versus more severe disease. Conversely, reducing the ppFEV1 threshold for performing LCI to <60% would increase the numbers of eligible patients but increase non-acceptable LCI values, with potential for patient and site frustration with the procedure.
Conclusions
Numerous attempts to demonstrate benefit with ENaC inhibition have failed [29], although a recent study with the ENaC antisense oligonucleotide ION-827359 in patients with CF has demonstrated a numerical dose-dependent increase in ppFEV1 after 4 weeks’ treatment, with a numerical 4.5% increase in the highest dose group versus placebo [45]. However, on balance, the potential of ENaC inhibition in patients with CF must be questioned. There is a clear medical need for further breakthroughs in CF targeting those patients not eligible for CFTR modulators, and for further normalisation of the status of patients who already receive CFTR modulators, with a drive toward simplification of treatment in this polytherapy disease. Whether this is through improvements in modulator approaches, channel approaches, treatment of inflammation, cure via gene therapy approaches, or other modalities, there continues to be a strong need for improvement in therapy.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary methods and tables ERJ-00746-2021.Supplement
Supplementary figure S1. (a) Change from baseline in unadjusted mean (±SE) trough ppFEV1 at weeks 1 and 4 of treatment – TS and (b) LCI after 4 weeks of treatment – N2MBWS. No patients in the BI 1265162 20 μg BID treatment group were included in the N2MBWS. The BI 1265162 50 μg and 100 μg BID treatment groups only had one patient each included in the N2MBWS; accordingly, no standard error could be calculated for these groups. BI: BI 1265162; BID: twice daily; LCI: lung clearance index; N2MBWS: N2 multiple breath washout set; ppFEV1: percent predicted forced expiratory volume in 1 s; SE: standard error; TS: treated set. ERJ-00746-2021.Figure_S1
Supplementary figure S2. Change from baseline in ppFEV1 after 4 weeks of treatment for individual patients, all dose groups – TS. *: additional patient visit data added after interim analysis. BI: BI 1265162; MMRM: mixed model for repeated measures; ppFEV1: percent predicted forced expiratory volume in 1 s; TS: treated set. ERJ-00746-2021.Figure_S2
Supplementary figure S3. Sensitivity analysis for change from baseline in ppFEV1 after 4 weeks of treatment for individual patients (MMRM) – TS. Data from visits were excluded based on AEs that might have affected pulmonary function tests, compliance, and unacceptable pulmonary function test quality at baseline and/or baseline condition considered as important protocol deviation. AE: adverse event; BI: BI 1265162; MMRM: mixed model for repeated measures; ppFEV1: percent predicted forced expiratory volume in 1 s; TS: treated set. ERJ-00746-2021.Figure_S3
Supplementary figure S4. ppFEV1 subgroup analyses (MMRM) of difference between BI 1265162 and placebo from baseline – TS. BMI: body mass index; CF: cystic fibrosis; CFTR: cystic fibrosis transmembrane conductance regulator; CI: confidence interval; MMRM: mixed model for repeated measures; ppFEV1: percent predicted forced expiratory volume in 1 s; TS: treated set. ERJ-00746-2021.Figure_S4
Supplementary figure S5. Change from baseline in LCI after 4 weeks of treatment for individual patients, final analysis – N2MBWS. BI: BI 1265162; LCI: lung clearance index; N2MBWS: N2 multiple breath washout set. ERJ-00746-2021.Figure_S5
Supplementary figure S6. Correlation analysis between change in CFQ-R Respiratory Domain score and change in ppFEV1 after 4 weeks of treatment. BI: BI 1265162; CFQ-R RD: Cystic Fibrosis Questionnaire – Revised Respiratory Domains; ppFEV1: percent predicted forced expiratory volume in 1 s. ERJ-00746-2021.Figure_S6
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00746-2021.Shareable
Acknowledgements
The authors would like to thank the study participants, study investigators and coordinators, the Cystic Fibrosis Foundation (CFF), the European Cystic Fibrosis Society (ECFS), the CFF Therapeutics Development Network, the CFF-DMC Chair and members, the ECFS-Clinical Trials Network, the ECFS Lung Clearance Index Core Facility (Clare Saunders and Christopher Short for test set-up, performance and analysis) and the Cystic Fibrosis Community Advisory Board in Europe. The authors would also like to thank the clinical study leader Anne-Caroline Picard for her operational excellence and Tina Luo for assistance with statistical analysis. Medical writing assistance, in the form of the preparation and revision of the manuscript, was supported financially by Boehringer Ingelheim and provided by Lee Kempster at MediTech Media (London, UK), under the authors’ conceptual direction and based on feedback from the authors. The study was supported by the National Institute of Health Research (NIHR) through the Imperial Biomedical Research Centre, the NHLI/Royal Brompton Clinical Research Facility and a Senior Investigator award (to J.C. Davies).
Footnotes
This article has supplementary material available from erj.ersjournals.com
This study is registered at ClinicalTrials.gov with identifier NCT04059094. To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to all relevant material, including participant-level clinical study data, and relevant material as needed by them to fulfil their role and obligations as authors under the ICMJE criteria. Furthermore, clinical study documents (e.g. study report, study protocol, statistical analysis plan) and participant clinical study data are available to be shared after publication of the primary manuscript in a peer-reviewed journal and if regulatory activities are complete and other criteria met per the BI Policy on Transparency and Publication of Clinical Study Data (https://trials.boehringer-ingelheim.com/transparency_policy.html). Prior to providing access, documents will be examined, and, if necessary, redacted and the data will be de-identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Clinical study reports and related clinical documents can be requested via this link: https://trials.boehringer-ingelheim.com/trial_results/clinical_submission_documents.html/. All such requests will be governed by a document sharing agreement. Bona fide, qualified scientific and medical researchers may request access to de-identified, analysable participant clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a data sharing agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Researchers should use https://clinicalstudydatarequest.com to request access to study data.
Conflict of interest: C.H. Goss reports grants from the Cystic Fibrosis Foundation, the European Commission and NIH (NHLBI, NIDDK and NCRR), during the conduct of the study; personal fees for grant review board work from Gilead Sciences, personal fees for data monitoring committee work from Novartis, grants from the NIH and FDA, non-financial support (reimbursement for travel and meeting attendance) and other (serving as trial lead with site contract) from Boehringer Ingelheim, personal fees for lectures from Vertex Pharmaceuticals, outside the submitted work.
Conflict of interest: I. Fajac reports grants and personal fees for consultancy from Boehringer, during the conduct of the study; grants and personal fees for consultancy from Proteostasis Therapeutics and Vertex Pharmaceuticals, personal fees for consultancy from Kither Biotech, outside the submitted work.
Conflict of interest: R. Jain reports grants and personal fees for consultancy from Vertex Pharmaceuticals, grants from the CF Foundation, Sound Pharma, Armata Pharmaceuticals, Corbus Pharmaceuticals and Genetech, grants and personal fees for advisory board work from Boehringer Ingelheim, outside the submitted work.
Conflict of interest: W. Seibold is an employee of Boehringer Ingelheim.
Conflict of interest: A. Gupta is an employee of Boehringer Ingelheim.
Conflict of interest: M-C. Hsu is a former employee of Boehringer Ingelheim (China) and current employee of Shanghai Junshi Biosciences Co Ltd.
Conflict of interest: S. Sutharsan reports personal fees for advisory board work from Vertex Pharmaceuticals, personal fees for lectures from Novartis, outside the submitted work.
Conflict of interest: J.C. Davies reports other (advisory board and clinical trial lead) from Algipharma AS, Bayer AG, Galapagos NV and Proteostasis Therapeutics, Inc., other (advisory board) from Boehringer Ingelheim Pharma GmbH & Co. KG, Nivalis Therapeutics, Inc., Raptor Pharmaceuticals, Inc., Enterprise, Novartis, ProQR Therapeutics III BV, Pulmocide and Flatley, other (advisory board and trial design assistance) from ImevaX GmbH and ProQR Therapeutics III BV, other (advisory board and national co-ordinator/global co-investigator) from Vertex Pharmaceuticals (Europe) Limited, grants from the CF Trust, other (educational activities) from Teva, outside the submitted work.
Conflict of interest: M.A. Mall reports grants, personal fees for advisory board work and non-financial support (travel expenses) from Boehringer Ingelheim, during the conduct of the study; personal fees for advisory board work, consultancy and lectures from Boehringer Ingelheim, personal fees for advisory board work and consultancy from Arrowhead Pharmaceuticals, Santhera, Enterprise Therapeutics, Antabio and Kither Biotech, grants and personal fees for advisory board work, consultancy and lectures from Vertex Pharmaceuticals, personal fees for consultancy from Galapagos and Sterna Biologicals, personal fees for lectures from Celtaxys, outside the submitted work.
Support statement: This work was supported by Boehringer Ingelheim. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received March 12, 2021.
- Accepted June 19, 2021.
- Copyright ©The authors 2022.
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org