



Plasma proteins elevated in severe asthma despite oral steroid use and unrelated to Type-2 inflammation
- Maria Sparreman Mikus1,2⇑,
- Johan Kolmert2,3,
- Lars I. Andersson2,3,
- Jörgen Östling4,
- Richard G. Knowles5,
- Cristina Gómez2,3,
- Magnus Ericsson6,
- John-Olof Thörngren6,
- Payam Emami Khoonsari7,
- Barbro Dahlén2,3,8,
- Maciej Kupczyk2,3,9,
- Bertrand De Meulder10,
- Charles Auffray10,
- Per S. Bakke11,
- Bianca Beghe12,
- Elisabeth H. Bel13,
- Massimo Caruso14,
- Pascal Chanez15,
- Bo Chawes16,
- Stephen J. Fowler17,
- Mina Gaga18,
- Thomas Geiser19,
- Mark Gjomarkaj20,
- Ildikó Horváth21,
- Peter H. Howarth22,
- Sebastian L. Johnston23,
- Guy Joos24,25,
- Norbert Krug26,
- Paolo Montuschi27,
- Jacek Musial28,
- Ewa Niżankowska-Mogilnicka28,
- Henric K. Olsson29,
- Alberto Papi30,
- Klaus F. Rabe31,
- Thomas Sandström32,
- Dominick E. Shaw33,
- Nikolaos M. Siafakas34,
- Mathias Uhlén1,35,
- John H. Riley36,
- Stewart Bates36,
- Roelinde J.M. Middelveld2,3,
- Craig E. Wheelock3,37,38,
- Kian Fan Chung23,
- Ian M. Adcock23,
- Peter J. Sterk13,
- Ratko Djukanovic22,
- Peter Nilsson1,
- Sven-Erik Dahlén2,3,38 and
- Anna James2,3
- on behalf of the U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Disease outcome) Study Group and the BIOAIR (Longitudinal Assessment of Clinical Course and Biomarkers in Severe Chronic Airway Disease) Consortium39
- 1Dept of Protein Science, KTH Royal Institute of Technology, SciLifeLab, Stockholm, Sweden
- 2Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
- 3Centre for Allergy Research, Karolinska Institutet, Stockholm, Sweden
- 4PEXA AB, Gothenburg, Sweden
- 5Knowles Consulting, Stevenage Bioscience Catalyst, Stevenage, UK
- 6Dept of Laboratory Medicine, Karolinska University Hospital Huddinge, Stockholm, Sweden
- 7Dept of Biochemistry and Biophysics, National Bioinformatics Infrastructure Sweden, Science for Life Laboratory, Solna, Sweden
- 8Dept of Medicine, Karolinska University Hospital Huddinge, Stockholm, Sweden
- 9Dept of Internal Medicine, Asthma and Allergy, Medical University of Lodz, University of Lodz, Lodz, Poland
- 10European Institute for Systems Biology and Medicine, Lyon, France
- 11Dept of Clinical Science, University of Bergen, Bergen, Norway
- 12Dept of Medical and Surgical Sciences, University of Modena and Reggio Emilia, Modena, Italy
- 13Dept of Respiratory Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands
- 14Dept of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy
- 15Assistance Publique des Hôpitaux de Marseille, Clinique des Bronches, Allergies et Sommeil, Aix Marseille Université, Marseille, France
- 16COPSAC, Copenhagen Prospective Studies on Asthma in Childhood, Herlev and Gentofte Hospital, University of Copenhagen, Copenhagen, Denmark
- 17Division of Infection, Immunity and Respiratory Medicine, School of Biological Sciences, The University of Manchester; Manchester Academic Health Science Centre and NIHR Manchester Biomedical Research Centre, Manchester University Hospitals NHS Foundation Trust, Manchester, UK
- 18Respiratory Medicine Dept and Asthma Centre, Athens Chest Hospital “Sotiria”, University of Athens, Athens, Greece
- 19Dept for Pulmonary Medicine, University Hospital and University of Bern, Bern, Switzerland
- 20Institute for Research and Biomedical Innovation, Italian National Research Council, Palermo, Italy
- 21Dept of Pulmonology, Semmelweis University, Budapest, Hungary
- 22NIHR Southampton Biomedical Research Centre, University Hospital Southampton NHS Foundation Trust, and Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton, UK
- 23National Heart and Lung Institute, Imperial College London, London, UK
- 24Dept of Internal Medicine and Pediatrics, Ghent University, Ghent, Belgium
- 25Dept of Respiratory Medicine, Ghent University Hospital, Ghent, Belgium
- 26Fraunhofer Institute for Toxicology and Experimental Medicine, Hannover, Germany
- 27Dept of Pharmacology, Università Cattolica del Sacro Cuore, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy
- 28Dept of Internal Medicine, Jagiellonian University Medical College, Krakow, Poland
- 29Translational Science and Experimental Medicine, Research and Early Development, Respiratory and Immunology, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
- 30Division of lnternal and Cardiorespiratory Medicine, University of Ferrara, Ferrara, Italy
- 31Dept of Internal Medicine, Christian Albrechts University Kiel, Kiel, Germany
- 32Dept of Public Health and Clinical Medicine, Umeå University, Umeå, Sweden
- 33Respiratory Research Unit, University of Nottingham, Nottingham, UK
- 34Dept of Thoracic Medicine, Medical School, University of Crete, Heraklion, Crete, Greece
- 35Dept of Neuroscience, Karolinska Institutet, Stockholm, Sweden
- 36Respiratory Therapeutic Unit, GlaxoSmithKline, London, UK
- 37Dept of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden
- 38Dept of Respiratory Medicine and Allergy, Karolinska University Hospital, Stockholm, Sweden
- 39The collaborating members of the U-BIOPRED and the BIOAIR study groups are listed in the Acknowledgements section
- Corresponding author: Maria Sparreman Mikus (maria.mikus{at}ki.se)
Abstract
Rationale Asthma phenotyping requires novel biomarker discovery.
Objectives To identify plasma biomarkers associated with asthma phenotypes by application of a new proteomic panel to samples from two well-characterised cohorts of severe (SA) and mild-to-moderate (MMA) asthmatics, COPD subjects and healthy controls (HCs).
Methods An antibody-based array targeting 177 proteins predominantly involved in pathways relevant to inflammation, lipid metabolism, signal transduction and extracellular matrix was applied to plasma from 525 asthmatics and HCs in the U-BIOPRED cohort, and 142 subjects with asthma and COPD from the validation cohort BIOAIR. Effects of oral corticosteroids (OCS) were determined by a 2-week, placebo-controlled OCS trial in BIOAIR, and confirmed by relation to objective OCS measures in U-BIOPRED.
Results In U-BIOPRED, 110 proteins were significantly different, mostly elevated, in SA compared to MMA and HCs. 10 proteins were elevated in SA versus MMA in both U-BIOPRED and BIOAIR (alpha-1-antichymotrypsin, apolipoprotein-E, complement component 9, complement factor I, macrophage inflammatory protein-3, interleukin-6, sphingomyelin phosphodiesterase 3, TNF receptor superfamily member 11a, transforming growth factor-β and glutathione S-transferase). OCS treatment decreased most proteins, yet differences between SA and MMA remained following correction for OCS use. Consensus clustering of U-BIOPRED protein data yielded six clusters associated with asthma control, quality of life, blood neutrophils, high-sensitivity C-reactive protein and body mass index, but not Type-2 inflammatory biomarkers. The mast cell specific enzyme carboxypeptidase A3 was one major contributor to cluster differentiation.
Conclusions The plasma proteomic panel revealed previously unexplored yet potentially useful Type-2-independent biomarkers and validated several proteins with established involvement in the pathophysiology of SA.
Abstract
Application of new proteomic panel in two established European asthma cohorts identifies plasma proteins associated with disease severity independently of Type-2 inflammation, suggesting potentially useful novel biomarkers and therapeutic targets. https://bit.ly/3jtTq5m
Introduction
Asthma is a prevalent chronic inflammatory disease with many different phenotypes sharing common clinical manifestations of episodic breathlessness, wheezing, cough, airflow obstruction (usually reversible) and airway hyperresponsiveness [1, 2]. The underpinning pathobiology may however be widely different. As individuals respond differently to treatments targeting specific pathways, better predictive biomarkers are required to improve patient selection, guide treatment and monitor responses. Currently available Type-2 asthma biomarkers, such as blood eosinophils, total serum immunoglobulin E (IgE) or fraction of exhaled nitric oxide (FeNO), do not adequately enable endotyping, which is required for personalised treatment [1]. Another major clinical challenge is the identification of biomarkers reflecting non-Type-2 asthma, which is often more severe and lacking effective treatments [3, 4].
With the overall aim being biomarker discovery, the ChAMP (Centre for Allergy Research highlights Asthma Markers of Phenotype) consortium developed an affinity proteomics panel focused on proteins with potential involvement in airway or systemic inflammation. Protein selection was based on literature reviews, database searches and recent research findings. The four main biological processes reflected by the proteins were 1) immune response, 2) lipid mediator pathways (predominantly sphingolipids), 3) signal transduction and 4) extracellular matrix (figure 1). The panel was enriched in non-Type-2-related proteins to address the unmet clinical need in this particular subgroup. Specifically, our objective was to examine plasma protein associations with asthma severity and oral corticosteroid (OCS) treatment. Furthermore, we aimed to test the hypothesis that specific plasma protein profiles can identify unique molecular subgroups of asthma patients.

Study overview. Two independent cohorts, U-BIOPRED and BIOAIR, were investigated in this study. In a first screening, the U-BIOPRED cohort including 525 baseline plasma samples from 525 subjects was profiled using antibody bead arrays detecting 177 proteins with 377 antibodies. In the validation stage, the same array was used to profile the BIOAIR cohort comprising 351 plasma samples from 142 subjects. The BIOAIR cohort included a double-blind placebo-controlled oral corticosteroid intervention trial where the placebo group received additional open steroid treatment. These samples were used to study the influence of steroids on plasma protein levels. Asthmatic subjects from the U-BIOPRED cohort were used to identify potential phenotypes using consensus clustering of protein profiles. COPD: chronic obstructive pulmonary disease; HC: healthy non-smoking controls; MMA: non-smokers with mild-to-moderate asthma; SAn: non-smokers with severe asthma; SAs/ex: smokers or ex-smokers with severe asthma.
Initially, subjects with mild-to-moderate (MMA) or severe (SA) asthma, and healthy controls (HCs) from the multicentre investigation U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Disease outcome) were screened [5]. The discoveries in U-BIOPRED were then validated in a second cohort, BIOAIR (Longitudinal Assessment of Clinical Course and BIOmarkers in Severe Chronic AIRway Disease) [6], where samples were obtained from subjects with MMA or SA or COPD, before and after a placebo-controlled intervention with oral prednisolone. This permitted comparisons between the protein profiles of subjects with SA and COPD, distinct respiratory disorders that may show certain overlapping characteristics and, importantly, an assessment of the pharmacological effect of oral glucocorticosteroids on the plasma proteins measured. The study design and main aims are shown in figure 1.
Methods
Subjects
The discovery cohort included baseline EDTA plasma samples from U-BIOPRED, a prospective cohort study of SA phenotypes (Clin.Trial.Gov NCT01976767) [5]. Of 525 included subjects, 263 were non-smokers with SA (SAn), 95 current or ex-smokers with SA (SAs/ex), 76 non-smokers with MMA and 91 HCs (figure 1). Subject characteristics and data availability are summarised in table 1 and table S1.
Demographic and clinical characteristics of study subjects
The validation cohort included 142 subjects from the BIOAIR study (Clin.Trial.Gov NCT00555607) [6]. After a 4-week treatment optimisation period, subjects underwent a 2-week, double-blind placebo-controlled OCS intervention (0.5 mg·kg−1·day−1 prednisolone) added to regular treatment, followed by an identical open OCS treatment of the placebo group only (figure 1). Heparin plasma samples from 58 SAn, 48 MMA and 36 patients with COPD, before and after the intervention, were analysed. Subject characteristics and data availability are shown in table 1 and table S1.
Further information regarding the study design of each cohort and diagnostic criteria for each subject group are presented in the supplementary material. Both studies were approved by the ethics committees of each participating clinical institution and participants provided written informed consent.
Panel design and suspension bead array protein profiling
Detailed information regarding the protein panel of 177 proteins (table S2) and the antibody bead array methodology is provided in the supplementary material. Briefly, this in-house developed array-based affinity proteomics method utilises antibodies coupled to magnetic colour-coded beads (MagPlex, Luminex Corp., Austin, TX, USA) to create a multiplex analysis platform [7, 8]. Measurements were performed using FlexMAP3D instruments (Luminex Corp.) and reported as relative fluorescent intensity values.
Statistical analysis
Statistical analysis and visualisation were performed in R [9, 10]. Non-parametric Kruskal–Wallis (multiple groups), Wilcoxon rank-sum (pairwise group) and Wilcoxon signed-rank test (paired) tests were used for comparisons of continuous variables. All reported p-values for the proteins were adjusted for multiple testing using the Benjamini and Hochberg [11] method and controlling the false discovery rate (FDR) at 5%.
Unsupervised consensus cluster analysis was used to identify potential subgroups of asthma patients, a process described in more detail in the supplementary material. Briefly, a reduced set of variables (n=139) was selected for consensus cluster analysis of log2 transformed and z-scored intensity signals for each antibody, performed using the “ConsensusClusterPlus” package in R [12, 13]. The Euclidean distance measure was used to describe similarity between subjects and the partitioning around the medoids algorithm was used for clustering. For model validation, clustering was repeated 1000 times, randomly removing 10% of subjects at each iteration. The cluster stability of models with two to 10 clusters was evaluated by the lowest proportion of ambiguously clustered subjects [14] and the lowest deviation of ideal stability [15]. A six-cluster model had slightly lower stability compared to 10 (table S8), but demonstrated greater consistency across all clusters (figure S2e) and is therefore presented.
To identify proteins important for classification of the six cluster groups, the Boruta algorithm (R package “Boruta”) [16], a wrapper built around the random forest classification method, was used. For more details, see the supplementary material.
Results
Screening for asthma-associated proteins in U-BIOPRED
Plasma profiling revealed 110 proteins (measured by 139 antibodies) that showed significantly different levels between subject groups in the U-BIOPRED cohort (table S4). The top 21 proteins, all with p-values lower than 10−10, are shown in table 2. The most significant differences were consistently identified between SAn or SAs/ex, and MMA or HC (figure 2a). No proteins were different between the non-smoking and smoking SA groups (SAn and SAs/ex), nor between MMA and HC (figure 2a). The overlap of proteins showing significant differences in the pairwise group comparisons are shown in figure S1a.
Plasma proteins significantly different between subject groups in the U-BIOPRED cohort

a) Volcano plots of pairwise group comparisons in U-BIOPRED highlight multiple proteins elevated in subjects with severe asthma, but not in mild-to-moderate. Each dot represents a protein measured by the antibody array. Dashed lines represent adjusted p-values <0.05. Highlighted in red are proteins significantly different in the respective pairwise group comparison as well as in the multiple group comparison (i.e. any of the 110 proteins). If multiple antibodies for the same target protein were significant, they all needed to show the same sign of log2 fold change. b) Group comparisons in U-BIOPRED when limited to subjects where oral corticosteroid (OCS) use was not reported and confirmed negative by urinary analysis (non-smokers with severe asthma (SAn) n=103, smokers or ex-smokers with severe asthma (SAs/ex) n=42, non-smokers with mild-to-moderate asthma (MMA) n=63, healthy control (HC) n=90). Highlighted in red are the proteins found to be significantly different in the respective pairwise group comparison based on all subjects (i.e. proteins that were significantly different in the respective comparisons in figure 2a). The majority of the proteins were still significantly different, seen by enrichment of red circles above the p-value threshold. (c) Analysis of steroid effect in U-BIOPRED. SAn and SAs/ex stratified by reported OCS use and OCS metabolite detection. OCS use defined as prescribed and confirmed positive in urine (SAn n=51, SAs/ex n=21) and no OCS defined as not prescribed and confirmed negative in urine (SAn n=103, SAs/ex n=42).
Validation of asthma severity-associated proteins in BIOAIR
In the validation cohort BIOAIR, between-group comparisons revealed significantly different plasma levels in 23 proteins (25 antibodies), see table S5. Of these, 15 proteins were altered between MMA and COPD (all elevated in COPD) and 13 between SAn and MMA (all elevated in SAn). No differences in protein levels were found between SAn and COPD. As shown in figure S1b, the majority of the significantly different proteins were unique to the SAn versus MMA, or the MMA versus COPD pairwise group comparison.
Comparing the results of U-BIOPRED and BIOAIR, 10 proteins were confirmed to be significantly different between SA and MMAin both cohorts (table 3). Alpha-1-antichymotrypsin (SERPINA3), apolipoprotein E (APOE), complement component 9 (C9), macrophage inflammatory protein-3 (CCL23 (MIP-3)), complement factor I (CFI), interleukin-6 (IL-6), sphingomyelin phosphodiesterase 3 (SMPD3), TNF receptor superfamily member 11a (TNFRSF11A (RANK)), transforming growth factor beta 1 (TGF-β1) and glutathione S-transferase P (GSTP1), were all elevated in SAn compared to MMA (figure 3).
Proteins successfully validated in both the U-BIOPRED and the BIOAIR cohort

Proteins validated in two independent cohorts. Boxplots of the 10 proteins showing significantly different plasma levels between non-smokers with severe asthma (SAn) and non-smokers with mild-to-moderate asthma (MMA) in both studied cohorts (adjusted p<0.05). The screening cohort U-BIOPRED and the validation cohort BIOAIR are shown side by side for each protein. COPD: chronic obstructive pulmonary disease; HC: healthy non-smoking control; MMA: non-smokers with mild-to-moderate asthma; SAn: non-smokers with severe asthma; SERPINA3: alpha-1-antichymotrypsin; APOE: apolipoprotein E; C9: complement component 9; CCL23: macrophage inflammatory protein 3; CFI: complement factor I; IL6: interleukin-6, SMPD3: sphingomyelin phosphodiesterase 3; TNFRSF11A: receptor activator of nuclear factor kappa B (RANK); TGF-β1: transforming growth factor beta-1; GSTP1: glutathione S-transferase P.
Influence of OCS on plasma protein levels
The highly significant differences in plasma protein levels observed between SA and MMA could be due to differences in disease mechanisms, pharmacological treatments, or both. We therefore used the BIOAIR cohort, where subjects underwent a 2-week controlled OCS trial with prednisolone, to examine the effect of glucocorticoid treatment on plasma protein levels. The majority of proteins were found to be affected by prednisolone, most of which were decreased (figure 4a and b). Among the 10 proteins that were elevated in SA in both BIOAIR and U-BIOPRED (figure 3), APOE, C9, MIP-3 and SERPINA3 all showed a significant change following oral steroid treatment. APOE and SERPINA3 were increased after OCS intake (figure 4c), whereas C9 and MIP-3 were decreased.
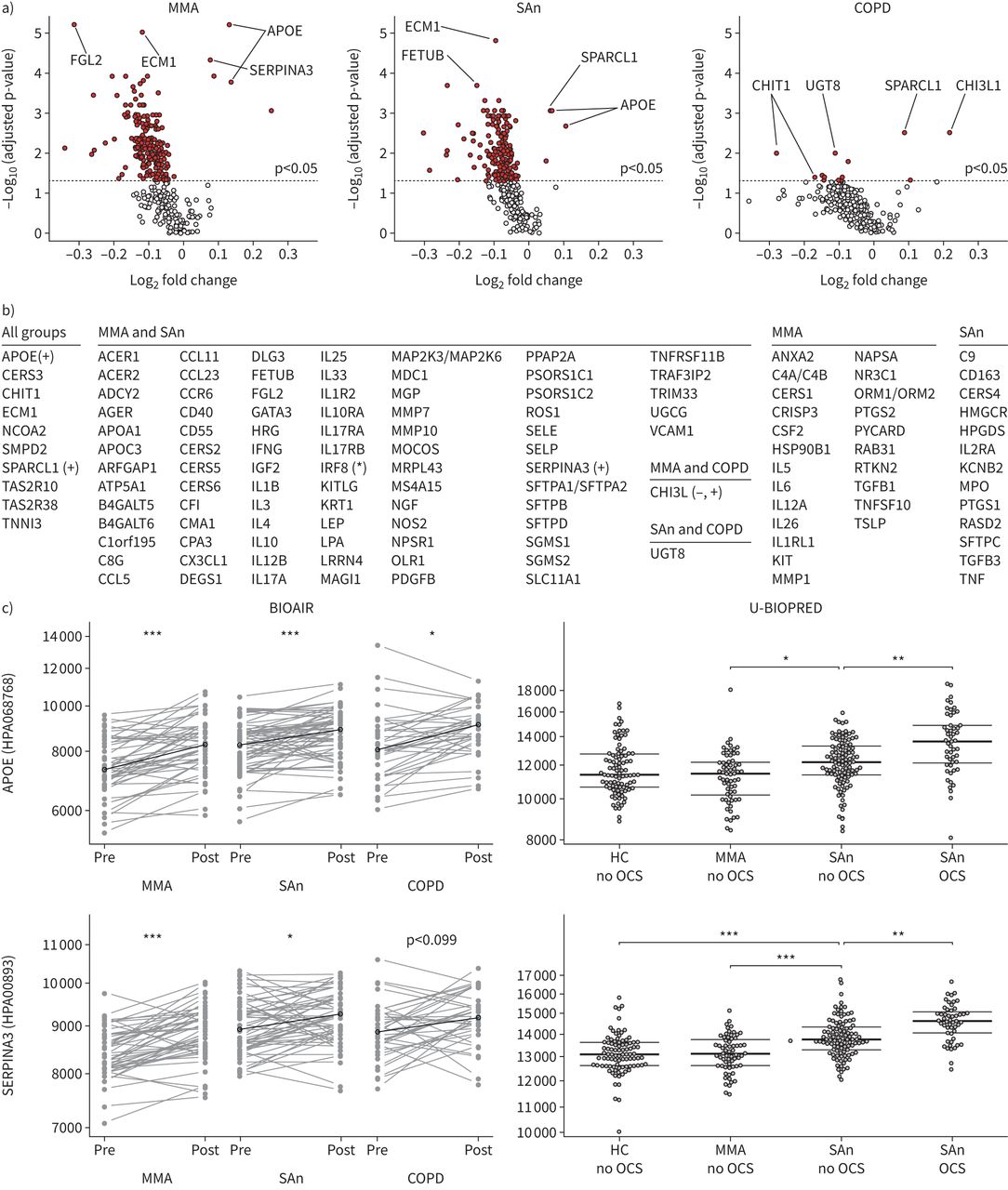
Influence of oral corticosteroids. a) Volcano plots of comparisons in BIOAIR show that multiple proteins were affected by the steroid treatment, with the majority being decreased. Each dot represents a protein measured by the antibody array, with significantly changing proteins highlighted in red (above the dashed lines representing adjusted p-values <0.05). The two most significant proteins on the decreasing and increasing side are labelled. Fold change calculated as log2 of the median of individual ratios (post/pre). b) List of proteins where the signal of at least one of the multiple antibodies targeting that protein in the array was affected by oral corticosteroid (OCS). Decreasing levels indicated by no mark and increasing levels marked with (+). Mixed directions provided by multiple antibodies are marked with (*) and mixed directions of effect in different subject groups are marked with (−,+). c) The plasma levels of apolipoprotein E (APOE) and alpha 1-antichymotrypsin (SERPINA3) increased after OCS in BIOAIR subject groups. In U-BIOPRED, APOE and SERPINA3 were associated with the severity of asthma as well as with the use of OCS among the severe asthmatics. Adjusted Wilcoxon signed-rank (BIOAIR) and rank-sum test (U-BIOPRED) p-values: *: p<0.05, **: p<0.01, ***: p<0.001. COPD: chronic obstructive pulmonary disease; HC: healthy non-smoking control; MMA: non-smokers with mild-to-moderate asthma; SAn: non-smokers with severe asthma. Full definitions of each protein name are listed in table S2.
Oral steroid-induced plasma protein changes found in BIOAIR were confirmed by combining prescription data and the objective measurement of urinary prednisolone metabolites in U-BIOPRED. Comparing levels between SAn reportedly taking and not taking OCS, as confirmed by urinary analysis, 20 proteins (21 antibodies) in U-BIOPRED were associated with oral steroid use (figure 2c and table S6). In line with BIOAIR results, APOE and SERPINA3 showed profiles that were both severity- and steroid-dependent, increasing from MMA to SAn not taking OCS, and further from SAn not taking OCS to SAn taking OCS (figure 4c). No proteins were significantly different when comparing the smaller groups of SAs/ex that used OCS and those who did not (figure 2c).
To take into account oral steroid effects, the multiple group comparison of SAn, SAs/ex, MMA and HC was repeated, limited to subjects reporting no current OCS use and in whom no urinary prednisolone metabolites were detected. Levels of 98 proteins (126 antibodies) remained significantly different between U-BIOPRED subject groups (figure 2b). Pairwise comparisons of these groups revealed a similar pattern as for the whole dataset, irrespective of OCS use (compare figure 2a with figure 2b). Again, there was no difference in levels between SAn and SAs/ex (data not shown). More importantly, the 10 replicated proteins remained elevated in SA (significant after FDR-adjustment: APOE, C9, MIP-3, CFI, SERPINA3 and RANK). A summary of all proteins that were significantly changed using these two approaches, including or excluding OCS users, is shown in table S7.
Identification of protein-driven subgroups of asthma
To identify subgroups or phenotypes of asthma, a protein-driven consensus clustering algorithm was applied to U-BIOPRED asthmatics. We clustered subjects based on their plasma protein profiles alone, restricted to the asthma-associated proteins identified in the univariate analysis (table S4). Evaluation of cluster models identified six robust clusters with high stability (table S8, figure 5a) and that were defined only by within-group similarities of included protein profiles. By applying a classification algorithm, it was possible to identify 108 profiles (85 unique proteins) confirmed as important for classification of the six clusters (figure S3). Although all proteins were relevant for classification, the three most contributing proteins are shown in figure 5b.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phenotypes of asthma. a) Six clusters were identified in a consensus clustering of asthmatic subjects (n=434, mild-to-moderate or severe asthma) from the U-BIOPRED cohort. The clustering was driven by the profiles of 110 proteins (139 antibodies). b) The three proteins CPA3 (carboxypeptidase A3), TRIM33 (tripartite motif containing protein 33) and TRAF3IP2 (TRAF3 interacting protein 2) with the greatest importance for cluster classification (for ranked importance of all proteins, see figure S3) and a selection of variables with a significant association with clusters. Boxes show the first quartile, median and third quartile. p-values are from the Kruskal–Wallis test. ACQ-7: Asthma Control Questionnaire-average of seven; AQLQ: Asthma Quality of Life Questionnaire–average; BMI: body mass index; FEV1: forced expiratory volume in 1 s; HC: non-smoking healthy controls; hsCRP: high-sensitivity C-reactive protein; tetranor-PGDM; 9α-hydroxy-11,15-dioxo-13,14-dihydro-2,3,4,5-tetranor-prostan-1,20-dioic acid.
A pattern was observed among the clusters when sorted by median plasma protein levels. Most commonly (63% of 139 antibody profiles), the highest levels were found in cluster 1, followed in decreasing order by clusters 6, 4, 3, 2 and 5. Decreasing protein plasma levels across the clusters were also associated with signs of decreasing disease severity (table S9). Although subjects with SA and MMA were found in all clusters, those with SA were relatively more prevalent in clusters 1, 4 and 6 (more than 90% per cluster) and subjects with MMA were relatively more prevalent in clusters 2, 3 and 5 (21%–37% of subjects per cluster) which contained 78% of all MMA subjects.
The clusters aggregated subjects with similar clinical outcomes (table S9). In cluster 1, subjects experienced low lung function, the most uncontrolled asthma, the lowest quality of life and the most frequent exacerbations. These subjects had the highest body mass index (BMI) (84% overweight, 60% obese) and evidence of systemic inflammation reflected by the most elevated high-sensitivity CRP (hsCRP) and highest blood neutrophils. They also showed signs of increased mast cell activation as reflected by elevated urinary tetranor-prostan-1,20-dioic acid (tetranor-PGDM). Conversely, cluster 5 included the youngest subjects with the best lung function, best asthma control, earliest onset of asthma and lowest BMI. These subjects also exhibited the lowest blood neutrophil numbers, lowest hsCRP and near-normal urinary tetranor-PGDM concentration. A selection of clinical and biochemical variables associated with the clusters is shown in figure 5b.
Parameters that were not different among the clusters included gender distribution and objective evidence of OCS use. Indices of Type-2 inflammation, including blood and sputum eosinophils, FeNO, circulating periostin, total serum IgE and prevalence of atopy, were also not significantly different (table S9).
Proteins associated with Type-2-high asthma
To examine whether proteins in the array were altered in subjects with Type-2-high compared to Type-2-low inflammation, we examined the U-BIOPRED data in relation to two recently published strategies for defining Type-2 asthma. Dividing asthma subjects according to a composite Type-2 biomarker score based on blood eosinophils, FeNO and serum periostin as described by the Refractory Asthma Stratification Programme (RASP) [17], was not associated with any significant differences in protein levels. In U-BIOPRED, we have recently published that high urinary leukotriene E4 (LTE4) levels have a strong Type-2 association [18]. In the current investigation, only seven of the proteins in the panel were associated with LTE4 levels (table S10). Taken together, this analysis confirms that the protein panel is suitable for its purpose to identify Type-2-independent signals.
Discussion
Using a high-throughput, array-based protein profiling technique to simultaneously analyse 177 selected plasma proteins in 667 subjects from two established European asthma cohorts, we identified both known and previously unknown candidate proteins associated with asthma severity. Despite observed effects of OCS on multiple proteins, associations with asthma severity remained for most proteins following adjustment for OCS use, a confounding factor often overlooked in biomarker discovery efforts. Furthermore, the patterns of protein profiles grouped asthmatics into clusters with clinically meaningful differences that were independent of Type-2 inflammation.
Among the 110 proteins significantly altered in severe compared to milder disease, many were known to be involved in regulation of immunological pathways. For example, the chitinases (CHI3L1 (YKL-40) and CHIT1 (chitotriosidase)) were elevated in both SA groups irrespective of smoking status compared with HC, in line with previous data [19]. Multiple inflammatory cytokines associated with the pathobiology of SA were also differentially abundant, including IL-4, IL-6, IL-10, IL-13, IL-17A, IL-26 and tumour necrosis factor-α [20, 21]. Severe asthmatics also showed increased levels of surfactant proteins, such as surfactant protein D, a pattern-recognition molecule involved in innate immune responses with antimicrobial activity [22]. Increased plasma levels are in line with previous findings [23], presumably reflecting increased epithelial damage and permeability within the peripheral airways.
Of the lipid mediator pathways, our findings suggest involvement of sphingolipids in asthma pathogenesis as multiple enzymes of sphingolipid metabolism were elevated in SA including ceramide synthases, sphingosine kinases and sphingomyelin phosphodiesterases. Components of the sphingolipid pathways, which have been shown to be involved in asthma and linked to disease severity [24, 25], represent potential therapeutic targets [26]. Several genome-wide association studies have also linked polymorphisms in the 17q21 locus where the orosomucoid 1-like 3 (ORMDL3) gene resides to both childhood and adult asthma [27, 28]. ORMDL3 is a regulator of sphingolipid synthesis and genetic variants lead to increased expression as well as decreased sphingolipid de novo synthesis in children with asthma [29]. Certain eicosanoid pathway enzymes were also elevated in SA such as haematopoietic prostaglandin D synthase and cyclooxygenase-1 which are involved in many processes, in particular mast cell activation [30].
Further proteins found to be increased in severe compared to mild asthma included members of the complement and coagulation cascades, confirming recent reports [31–33], as well as metabolic factors such as insulin and leptin. Proteins related to oxidative stress, including superoxide dismutase and GSTP1, were also elevated. These proteins have previously been associated with air pollution and mild asthma [34], but their pathobiological role in SA is largely unknown.
The follow-up investigation in the BIOAIR cohort replicated some of the findings with 10 proteins being significantly increased in SA compared to MMA in both cohorts. Many more proteins also followed the same trends as observed in U-BIOPRED, but did not reach statistical significance, possibly due to the smaller size of BIOAIR. It is also possible that this number may have been greater if plasma had been collected in exactly the same way in both studies (i.e. both heparin, or both EDTA plasma samples). However, the studies were planned and conducted at different times by different consortia, and therefore procedures were not identical. This study nevertheless demonstrates the complementary power of BIOAIR and U-BIOPRED, despite minor differences in methodology.
Among replicated findings were proteins with known involvement in airway fibrosis and remodelling such as TGF-β1 [35] and SERPINA3. The latter might be of particular relevance in COPD, where plasma levels have been found to be elevated [36]. IL-6, which may play a role in the pathobiology of a specific, exacerbation-prone, asthma sub-phenotype [37], was also increased in SA patients. Interestingly, plasma MIP-3 was elevated, consistent with a strong association of the gene encoding for MIP-3 with suboptimal asthma control [38]. Indeed, in the cluster analysis, MIP-3 showed the highest levels in the cluster with the worst Asthma Control Questionnaire and Asthma Quality of Life Questionnaire scores, and was among the most significant proteins differentiating between clusters. In the context of asthma, APOE has received attention due to its ability to suppress airway inflammation, and one may speculate that the increased levels observed in SA represent a protective function [39]. However, apolipoproteins may also be affected by statin therapy and for this reason we investigated whether APOE differed between MMA and SA following removal of patients with cardiovascular comorbidities. In both U-BIOPRED and BIOAIR, APOE remained significantly greater in SA than MMA, data not shown.
One particularly novel aspect of this study was the exploration of plasma protein profiles for the clustering of asthmatics into consensus groups beyond their clinical diagnosis. This yielded six clusters of subjects with differences in clinical and biochemical parameters. Clusters sorted by decreasing median plasma levels of the studied proteins were associated with phenotypes of decreasing severity. In summary, we observed associations between the cluster groups and age, age of asthma onset, BMI, forced expiratory volume in 1 s, exacerbations, blood neutrophils, serum hsCRP, asthma control and quality of life. However, no significant differences across clusters were observed in measures associated with Type-2 inflammation, including blood and sputum eosinophils, FeNO, total serum IgE, serum periostin, RASP Type-2 score or atopy. Along with the relative lack of differences in protein levels observed when dividing asthmatics into Type-2-high and Type-2-low subgroups, these findings confirm that the protein panel was fit for the purpose of identifying Type-2-independent signals. Furthermore, the proportion of subjects with detectable urinary levels of prednisolone metabolites was similar among the clusters, suggesting that OCS use was not a factor driving the observed cluster differences.
Plasma proteins that were particularly important for cluster classification included carboxypeptidase A3 (CPA3, a mast cell specific carboxypeptidase stored in secretory granules), tripartite motif containing protein 33 and TRAF3 interacting protein 2. In fact, the cluster-driven increase in CPA3 was mimicked by elevated urinary tetranor-PGDM, the major urinary metabolite of the main mast cell prostanoid prostaglandin D2 (PGD2). Collectively, this evidence suggests a pivotal role for mast cell activation in Type-2-independent SA. Interestingly, and in contrast to these findings, we previously showed that PGD2 is associated with Type-2 inflammation [18]. Thus, taken together our findings indicate that mast cells are diametric immune cells with a wide-spread involvement in both Type-2 and non-Type-2 inflammation.
Several new biological treatments have been approved for insufficiently controlled Type-2 asthma, although what currently represents a major unmet medical need is improved therapy for the subgroup of asthmatics with little or no Type-2 inflammation [1, 2, 40]. Interestingly, the most striking differences in clinical characteristics between clusters included the high BMI observed in cluster 1, a group which also had the highest blood neutrophils, highest serum CRP, worst quality of life and most exacerbations, possibly reflecting a non-Type-2 asthma group. Serum CRP has been suggested as a biomarker for neutrophilic asthma [41] and, accordingly, a neutrophilic inflammatory phenotype of asthma was associated with increased systemic inflammation, as reflected by elevated serum CRP levels compared with non-neutrophilic asthma [42]. Our findings may therefore provide a set of new, potential Type-2-independent biomarkers.
A major strength of the current investigation is the use of two cohorts of well-characterised asthmatic subjects, enabling validation of findings in an independent population and providing a comparison with COPD. Incidentally, SA and COPD could not be differentiated based on levels of the measured proteins in plasma, providing support for “the Dutch hypothesis” [43] and common molecular features in severe obstructive inflammatory airway diseases. One may speculate that the lack of difference between SA and COPD in BIOAIR could be due to the presence of patients with asthma/COPD overlap in either of these groups. This is, however, unlikely as the inclusion/exclusion criteria of the BIOAIR study were designed to reduce possible overlap between diagnoses. An exclusion factor for SA was current smoking (>10 cigarettes per day) and a history of greater than five pack years. In fact, the median number of pack years in the BIOAIR SA group was 0, and reversibility was also significantly higher, as expected, in SA compared to COPD [44]. An exclusion factor for COPD was diagnosed asthma or allergy and, furthermore, COPD patients were required to be current or ex-smokers with a history of >15 pack years.
The two study designs were also complementary with U-BIOPRED providing data from a larger, observational, cross-sectional study. BIOAIR, on the other hand, although smaller, included two interventions: first 4 weeks of standardised treatment, which reduced biological variability due to differences in therapy, and then a placebo-controlled OCS trial. A further strength was the design of the array by the ChAMP consortium, based on multiple hypotheses concerning signalling pathways of relevance to asthma, airway inflammation and immune-mediated diseases. The outcome was successful as the majority of proteins indeed displayed differential levels between disease severity and treatment groups. Finally, a methodological advantage of the assay itself was that, for several analytes, multiple antibodies targeting the same protein were used, often showing supporting profiles (figure S4).
A further advantage of the current investigation is that the analysis was based on the readily available plasma matrix, supporting future development of less invasive tools for molecular asthma phenotyping at the point of care. The detection of elevated airway proteins, such as surfactant proteins, in plasma from subjects with SA and COPD, suggesting increased leakage in severe disease, also confirms that disease processes in the lung tissue may be reflected in plasma. Generally, the findings suggest a greater systemic impact of SA compared to MMA as protein levels were similar in mild asthma and HCs.
Possible effects of therapy are often overlooked in biomarker discovery efforts, but the BIOAIR oral prednisolone intervention enabled a thorough assessment of the effect of OCS therapy on plasma protein profiles. Results showed that levels of most proteins were reduced by OCS treatment. Interestingly, when U-BIOPRED subjects taking OCS were removed from analyses (figure 2b), the differences between MMA and SA were maintained for most proteins, although not for all, suggesting that plasma levels were affected both by disease severity and OCS treatment. Moreover, proteins that were altered by OCS in the non-smoking SA group did not change with OCS use in the smoking SA group where, in fact, no proteins changed (figure 2c). This may reflect the known decreased responsiveness to corticosteroids and differences in inflammatory biomarkers among smoking asthmatics [45, 46] and/or be a power issue. The SA groups in both U-BIOPRED and BIOAIR were taking higher doses of inhaled corticosteroids than the MMA groups, as per inclusion criteria, and it is important to note that inhaled corticosteroids could potentially also affect protein expression. Unfortunately, a limitation of the current investigation is that we were not able to examine this possibility as rigorously as for OCS.
In conclusion, the antibody array we developed identified multiple plasma proteins associated with asthma severity, both confirming the involvement of known proteins such as inflammatory cytokines, growth factors and chitinases, as well as suggesting novel targets for further investigation. For example, our findings advocate components of the sphingolipid pathway and complement cascades as well as mast cell related proteins as being worthy of more detailed future examinations. These discoveries were found to be independent of effects potentially related to OCS therapy. Furthermore, we showed that protein profiles, driven by amongst others a mast cell-specific carboxypeptidase, could be used to group asthmatic subjects into six clinically distinct clusters. Taken together, we present a platform that is able to suggest novel biomarker candidates for molecular phenotyping, particularly relevant to non-Type-2 asthma. The panel may also aid discovery of future pharmacotherapeutic targets by exposing previously unexplored pathways.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00142-2021.SUPPLEMENT
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00142-2021.Shareable
Acknowledgements
Support by NBIS (National Bioinformatics Infrastructure Sweden) is gratefully acknowledged. We also thank the entire staff of the Human Protein Atlas and the group of Affinity proteomics at SciLifeLab Stockholm for their great efforts. All the members of U-BIOPRED Study Group and the BIOAIR consortium are gratefully acknowledged.
Footnotes
U-BIOPRED Study Group members: Hassan Ahmed, David Balgoma, Aruna T. Bansal, Frédéric Baribaud, Jeanette Bigler, Bo Billing, Hans Bisgaard, Michael J. Boedigheimer, Klaus Bønnelykke, Joost Brandsma, Paul Brinkman, Enrica Bucchioni, Dominic Burg, Andrew Bush, Amphun Chaiboonchoe, Toni Checa, Chris H. Compton, Julie Corfield, Danen Cunoosamy, Arnaldo D'Amico, Rosalia Emma, Veit J. Erpenbeck, Damijan Erzen, Klaus Fichtner, Neil Fitch, Louise J. Fleming, Elena Formaggio, Urs Frey, Martina Gahlemann, Victoria Goss, Yi-Ke Guo, Simone Hashimoto, John Haughney, Gunilla Hedlin, Pieter-Paul W. Hekking, Tim Higenbottam, Jans M. Hohlfeld, Cecile T.J. Holweg, Alan J. Knox, Jon Konradsen, Nikolaos Lazarinis, Diane Lefaudeux, Tracy Li, Matthew J. Loza, Rene Lutter, Alexander Manta, Sarah Masefield, John G. Matthews, Alexander Mazein, Andrea Meiser, Montse Miralpeix, Nadia Mores, Clare S. Murray, David Myles, Shama Naz, Björn Nordlund, Laurie Pahus, Ioannis Pandis, Stelios Pavlidis, Anthony Postle, Pippa Powel, Navin Rao, Stacey Reinke, Amanda Roberts, Graham Roberts, Anthony Rowe, James P.R. Schofield, Wolfgang Seibold, Anna Selby, Ralf Sigmund, Florian Singer, Marcus Sjödin, Paul J. Skipp, Ana R. Sousa, Kai Sun, Bob Thornton, Mohib Uddin, Wim M. van Aalderen, Marleen van Geest, Jorgen Vestbo, Nadja H. Vissing, Arianne H. Wagener, Scott S. Wagers, Zsoka Weiszhart, Craig E. Wheelock, Åsa Wheelock, Susan J. Wilson and Valentyna Yasinska. BIOAIR consortium members: Guy G. Brusselle, Deborah A. Campbell, Marco Contoli, Katarina Damm, Isabelle de Rudder, Ingrid Delin, Catherine Devautour, Mariusz Duplaga, Marianne Eduards, Alexandra Ek, Tommy Ekström, Ewa Figiel, Flora Gaber, Stefanie Gauw, Agnieszka Gawlewicz-Mroczka, Daisy Gerding, Shushila Haque, Lorraine Hewitt, Pieter S. Hiemstra, Stephen T. Holgate, John Holloway, Aleksander Kania, Frank Kanniess, Östen Karlsson, Johan C. Kips, Maria Kumlin, Ann-Sofie Lantz, Nikos Lazarinis, Helgo Magnussen, Patrick Mallia, Ingrid Martling, Lahouari Meziane, Erasmia Oikonomidou, Marianne Olsson, Elisabetta Pace, Eva Papadopouli, Nikos Papadopoulos, Maria Plataki, Mirella Profita, Lovisa E. Reinius, Kai Richter, Douglas S. Robinson, Micaela Romagnoli, Katerina Samara, Vanessa Schelfhout, Maria Skedinger, Evangelia Stamataki, Anneke ten Brinke, Isabelle Vachier, Eva Wallén-Nielsen, Ilonka van Veen, Els Weersink, Susan J. Wilson, Valentyna Yasinska, Eleftherios Zervas and Bozena Ziolkowska-Graca.
This article has supplementary material available from erj.ersjournals.com
This article has an editorial commentary: https://doi.org/10.1183/13993003.02669-2021
Author contributions: Conception, design or clinical conduct of the study: R.G. Knowles, B. Dahlén, M. Kupczyk, C. Auffray, P.S. Bakke, B. Beghe, E.H. Bel, M. Caruso, P. Chanez, B. Chawes, S.J. Fowler, M. Gaga, T. Geiser, M. Gjomarkaj, I. Horváth, P.H. Howarth, S.L. Johnston, G. Joos, N. Krug, P. Montuschi, J. Musial, E. Niżankowska-Mogilnicka, H.K. Olsson, A. Papi, K.F. Rabe, T. Sandström, D.E. Shaw, N.M. Siafakas, M. Uhlen, J.H. Riley, S. Bates, R.J.M. Middelveld, C.E. Wheelock, K.F. Chung, I.M. Adcock, P.J. Sterk, R. Djukanovic, P. Nilsson and S-E. Dahlén. Acquisition, analysis, or interpretation of data: M. Sparreman Mikus, J. Kolmert, L.I. Andersson, J. Östling, C. Gómez, M. Ericsson, J-O. Thörngren, P. Emami Khoonsari, B. De Meulder, C.E. Wheelock, P. Nilsson, S-E. Dahlén and A. James. Drafting the work: M. Sparreman Mikus, J. Kolmert, L.I. Andersson, P. Nilsson, S-E. Dahlén and A. James. Revising it critically for important intellectual content and final approval of the manuscript version to be published: all authors. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: M. Sparreman Mikus, P. Nilsson, S-E. Dahlén and A. James.
Conflict of interest: M. Sparreman Mikus has nothing to disclose.
Conflict of interest: J. Kolmert reports personal fees from Gesynta Pharma AB.
Conflict of interest: L.I. Andersson has nothing to disclose.
Conflict of interest: J. Östling has nothing to disclose.
Conflict of interest: R.G. Knowles has nothing to disclose.
Conflict of interest: C. Gómez has nothing to disclose.
Conflict of interest: M. Ericsson has nothing to disclose.
Conflict of interest: J-O. Thörngren has nothing to disclose.
Conflict of interest: P. Emami Khoonsari has nothing to disclose.
Conflict of interest: B. Dahlén reports personal fees from AstraZeneca, Teva, Sanofi and grants from Novartis and GlaxoSmithKline outside the submitted work.
Conflict of interest: M. Kupczyk has nothing to disclose.
Conflict of interest: B. De Meulder report grants from the Innovative Medicines Initiative during the conduct of the study.
Conflict of interest: C. Auffray report grants from the Innovative Medicines Initiative during the conduct of the study.
Conflict of interest: P.S. Bakke reports personal fees from GlaxoSmithKline, AstraZeneca, Boehringer Ingelheim and Chiesi outside the submitted work.
Conflict of interest: B. Beghe reports personal fees from AstraZeneca, Boehringer Ingelheim, Menarini and GlaxoSmithKline outside the submitted work.
Conflict of interest: E.H. Bel reports grants and personal fees from GlaxoSmithKline, AstraZeneca, Novartis, TEVA, Sanofi/Regeneron, Chiesi, and Sterna outside the submitted work.
Conflict of interest: M. Caruso has nothing to disclose.
Conflict of interest: P. Chanez has nothing to disclose.
Conflict of interest: B. Chawes has nothing to disclose.
Conflict of interest: S.J. Fowler has nothing to disclose.
Conflict of interest: M. Gaga reports grants and personal fees from Novartis, Menarini, Merck Sharp & Dohme, BMS, Galapagos, and AstraZeneca outside the submitted work.
Conflict of interest: T. Geiser has nothing to disclose.
Conflict of interest: M. Gjomarkaj has nothing to disclose.
Conflict of interest: I. Horváth reports grants from EFPIA during the conduct of the study, and personal fees from AstraZeneca, GlaxoSmithKline, Novartis, Boehringer-Ingelheim, Sandoz, Teva and Chiesi outside the submitted work.
Conflict of interest: P.H. Howarth has nothing to disclose.
Conflict of interest: S.L. Johnston reports personal fees from Virtus Respiratory Research, Myelo Therapeutics GmbH, Concert Pharmaceuticals, Bayer, Synairgen, Novartis, Boehringer Ingelheim, Chiesi, Gerson Lehrman Group, resTORbio, Bioforce, Materia Medical Holdings, PrepBio Pharma, Pulmotect, Virion Health, Lallemand Pharma and AstraZeneca outside the submitted work. In addition, Sebastian L. Johnston also has three patents (Anti-virus therapy for respiratory diseases, UK patent application No. GB 0405634.7, Interferon-Beta for Anti-Virus Therapy for Respiratory Diseases, International Patent Application No. PCT/ GB05/50031 and Interferon Lambda therapy for the treatment of respiratory disease, UK patent application No.6779645.9).
Conflict of interest: G. Joos reports grants and personal fees from AstraZeneca, Bayer, Chiesi, Eureca vzw, GlaxoSmithKline and Teva outside the submitted work.
Conflict of interest: N. Krug has nothing to disclose.
Conflict of interest: P. Montuschi has nothing to disclose.
Conflict of interest: J. Musial has nothing to disclose.
Conflict of interest: E. Niżankowska-Mogilnicka has nothing to disclose.
Conflict of interest: H.K. Olsson reports other support from AstraZeneca outside the submitted work.
Conflict of interest: A. Papi reports grants, personal fees, non-financial support and others from GlaxoSmithKline, AstraZeneca, Boehringer Ingelheim, Chiesi, Teva, Mundipharma, Zambon, Novartis, Menarini, Sanofi/Regeneron, Roche, Fondazione Salvatore Maugeri, Chiesi and Edmond pharma outside the submitted work.
Conflict of interest: K.F. Rabe reports grants and personal fees from AstraZeneca, Boehringer Ingelheim, Sanofi Aventis, MERCK SHARP & DOHME, Novartis, Orion Cooperation, Berlin Chemie, Roche, Chiesi and grants for research from the Ministry of Education and Science, Germany.
Conflict of interest: T. Sandström has nothing to disclose.
Conflict of interest: D.E. Shaw reports personal fees and non-financial support from GlaxoSmithKline, Novartis and AstraZeneca outside the submitted work.
Conflict of interest: N.M. Siafakas has nothing to disclose.
Conflict of interest: M. Uhlen has nothing to disclose.
Conflict of interest: J.H. Riley is an employee and shareholder in GlaxoSmithKline.
Conflict of interest: S. Bates is an employee and shareholder in GlaxoSmithKline.
Conflict of interest: R.J.M. Middelveld reports grants from the Swedish Strategic Research Foundation, AstraZeneca, the Swedish Heart Lung Foundation and the Swedish Asthma and Allergy Association outside the submitted work.
Conflict of interest: C.E. Wheelock has nothing to disclose.
Conflict of interest: K.F. Chung reports grants and personal fees from GlaxoSmithKline, AstraZeneca, Novartis, Merck, Boehringer Ingelheim, Roche and Shionogi outside the submitted work.
Conflict of interest: I.M. Adcock has nothing to disclose.
Conflict of interest: P.J. Sterk reports grants to Amsterdam UMC from the public private Innovative Medicines Initiative (IMI) covered by the European Union (EU) and the European Federation of Pharmaceutical Industries and Associations (EFPIA) during the conduct of the study.
Conflict of interest: R. Djukanovic reports receiving fees for lectures at symposia organised by Novartis, GlaxoSmithKline, AstraZeneca and Teva, consultation fees from Teva and Novartis; he is a co-founder and current consultant and has shares in Synairgen.
Conflict of interest: P. Nilsson has nothing to disclose.
Conflict of interest: S-E. Dahlén reports personal fees from AstraZeneca, Cayman Chemicals, GlaxoSmithKline, Novartis, Regeneron, Sanofi and Teva outside the submitted work.
Conflict of interest: A. James has nothing to disclose.
Support statement: This study was supported by grants from the ChAMP (Centre for Allergy Research Highlights Asthma Markers of Phenotype) consortium which is funded by the Swedish Foundation for Strategic Research, the Karolinska Institutet, AstraZeneca and Science for Life Laboratory Joint Research Collaboration, and the Vårdal Foundation, the Swedish Heart-Lung Foundation, the Swedish MRC, the Stockholm County Council Research Funds (ALF), the Swedish Asthma and Allergy Association's Research Foundation, and Karolinska Institutet. This project has received funding from an Innovative Medicines Initiative (IMI) Joint Undertaking (JU) under grant agreement number 115010 (U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Diseases Outcomes)), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution. Additional support from IMI 2 JU under grant agreement number 831434 (3TR (Taxonomy, Treatment, Targets and Remission)). This JU receives the support from the European Union's Horizon 2020 research and innovation programme and EFPIA. The BIOAIR consortium was funded by the Fifth and Sixth Framework Programmes of the European Union, contract numbers: QLG1-CT-2000-01185 (BIOAIR) and FOOD-CT-2004-506378 (GA2LEN), and several national funding bodies. A. James and M. Kupczyk were supported by the Bernard Osher Initiative for Severe Asthma. P. Emami Khoonsari was financially supported by the Knut and Alice Wallenberg Foundation as part of the National Bioinformatics Infrastructure Sweden at SciLifeLab. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received January 15, 2021.
- Accepted June 24, 2021.
- Copyright ©The authors 2022.
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org