



Neutrophilic inflammation in asthma and defective epithelial translational control
- Abilash Ravi1,2,
- Saheli Chowdhury1,2,
- Annemiek Dijkhuis2,
- Peter I. Bonta1,
- Peter J. Sterk1 and
- René Lutter1,2⇑
- 1Amsterdam UMC, University of Amsterdam, Dept of Respiratory Medicine, Amsterdam, The Netherlands
- 2Amsterdam UMC, University of Amsterdam, Dept of Experimental Immunology, Amsterdam Infection and Immunity Institute, Amsterdam, The Netherlands
- René Lutter, Amsterdam University Medical Centers, University of Amsterdam, Depts Exp. Immunology/Respiratory Medicine, K0-150, P.O. Box 22700, Amsterdam, 1100 DE, The Netherlands. E-mail: r.lutter{at}amsterdamumc.nl
Abstract
Neutrophilic inflammation in asthma is associated with interleukin (IL)-17A, corticosteroid-insensitivity and bronchodilator-induced forced expiratory volume in 1 s (FEV1) reversibility. IL-17A synergises with tumour necrosis factor (TNF)-α in the production of the neutrophil chemokine CXCL-8 by primary bronchial epithelial cells (PBECs).
We hypothesised that local neutrophilic inflammation in asthma correlates with IL-17A and TNF-α-induced CXCL-8 production by PBECs from asthma patients.
PBECs from most asthma patients displayed an exaggerated CXCL-8 production in response to TNF-α and IL-17A, but not to TNF-α alone, and which was also insensitive to corticosteroids. This hyperresponsiveness of PBECs strongly correlated with CXCL-8 levels and neutrophil numbers in bronchoalveolar lavage from the corresponding patients, but not with that of eosinophils. In addition, this hyperresponsiveness also correlated with bronchodilator-induced FEV1 % reversibility. At the molecular level, epithelial hyperresponsiveness was associated with failure of the translational repressor T-cell internal antigen-1 related protein (TiAR) to translocate to the cytoplasm to halt CXCL-8 production, as confirmed by TiAR knockdown. This is in line with the finding that hyperresponsive PBECs also produced enhanced levels of other inflammatory mediators.
Hyperresponsive PBECs in asthma patients may underlie neutrophilic and corticosteroid-insensitive inflammation and a reduced FEV1, irrespective of eosinophilic inflammation. Normalising cytoplasmic translocation of TiAR is a potential therapeutic target in neutrophilic, corticosteroid-insensitive asthma.
Abstract
Defective translational control of neutrophil-driving cytokines in bronchial epithelium leads to exaggerated, corticosteroid-insensitive production in vitro and correlates with neutrophilic inflammation in airway lumen and FEV1 reversibility http://bit.ly/2VrHS2M
Introduction
Asthma is a heterogeneous disease characterised by chronic airway inflammation and airway hyperresponsiveness culminating in episodes of reversible airway obstruction. This heterogeneity is reflected by differences in pathology, clinical course and response to standard treatment [1, 2]. An understanding of molecular mechanisms underlying this heterogeneity may provide a rationale for targeted treatment of patients. This is exemplified by successful therapeutic interventions with anti-interleukin (IL)-5 [3, 4] and anti-IgE [5] in severe asthma patients with a marked eosinophilic inflammation [6] and type 2 T-helper cell (Th2)-high signature, respectively. Treatment strategies for asthma with a predominant neutrophilic inflammation are not available, which relates to the poor understanding of mechanisms that drive asthma pathogenesis in these patients.
IL-17A has been associated with neutrophilic inflammation [7, 8] and corticosteroid-insensitivity in allergic asthma [9], in severe asthma and in exacerbations of asthma [10–12]. In addition, IL-17A and neutrophilic inflammation have been linked to the post-bronchodilator forced expiratory volume in 1 s (FEV1) response [13, 14]. Among others, IL-17A targets stromal cells [7] such as primary bronchial epithelial cells (PBECs), which are among the most abundant cell type in the airways and which produce many inflammatory mediators relevant to asthma and therefore may drive airway inflammation [15]. In addition, IL-17A has been shown to decrease the responsiveness of CXCL-8 production by the bronchial epithelium to corticosteroids [16]. IL-17A synergises with pro-inflammatory stimuli such as tumour necrosis factor (TNF)-α in the production of CXCL-8 and IL-6 by attenuating degradation of CXCL8 and IL6 mRNA [17–19]. Previously, we have shown that CXCL8 and IL6 mRNA degradation is promoted by microRNA (miR)16 expression, which is attenuated by IL-17A [20]. Hence, we hypothesised that PBECs from asthma patients release more CXCL-8 and IL-6 in response to IL-17A and TNF-α than PBECs from healthy individuals, because of reduced CXCL8 and IL6 mRNA degradation.
PBECs collected by bronchial brushing were expanded and the production of mediators relevant to neutrophilic inflammation and other mediators were determined after in vitro stimulation of PBECs with IL-17A and TNF-α. Despite enhanced CXCL-8 and IL-6 responses and high miR16 levels in PBECs from most asthma patients, the half-lives of CXCL8 and IL6 mRNA were not different in PBECs from healthy controls. Instead we found a defective cytoplasmic translocation of the translational repressor T-cell internal antigen-1 related protein (TiAR) in PBECs from asthma patients, which related to corticosteroid insensitivity, in vivo neutrophilic inflammation and the post-bronchodilator FEV1 response.
Methods
Study design
Bronchial epithelial brushes were obtained from three registered trials, the RESOLVE (NCT1677) study with healthy and mild asthma patients, the MATERIAL (NTR01520051) study with mild asthma patients and TASMA (NCT02225392) study with severe asthma patients. After inclusion, bronchoalveolar lavage fluid (BALF) and bronchial brushes were collected by standard bronchoscopy procedures [21] on the same day. Lung function parameters, FEV1 % reversibility and methacholine challenge test (provocative concentration causing a 20% fall in FEV1 (PC20)) were performed 4 days prior to bronchoscopy.
Subjects
The study protocols were reviewed and approved by the ethical review committee and were in accordance with the declaration of Helsinki. All patients and controls provided written informed consent. The studies were conducted in one centre only, at the Department of Respiratory Medicine of the Academic Medical Center (Amsterdam, the Netherlands). Patients with mild allergic asthma (RESOLVE and MATERIAL studies) met the following criteria: a history of episodic chest symptoms, baseline FEV1 >80% predicted, airway responsiveness to methacholine (PC20 <9.8 mg·mL−1), skin prick test positive for at least one of 12 common aeroallergens. Patients were not allowed to use inhaled or systemic corticosteroids or treatment other than inhaled short-acting β2-agonists within 2 weeks prior to the start of the study. Healthy individuals from the RESOLVE study had a FEV1 >80% pred, PC20 >16 mg·mL−1, skin prick test negative for 12 common aeroallergens. All mild asthma patients and healthy controls were nonsmoking or had stopped smoking 12 months ago with ≤5 pack-years.
Patients with severe asthma (TASMA study), fulfilling the World Health Organization or modified Innovative Medicines Initiative criteria of severe refractory asthma were included [22, 23]. Patients using inhaled corticosteroids at a dosage ≥500 μg fluticasone equivalent per day and long acting β2-agonist at a dosage of ≥100 μg per day salmeterol dose aerosol or equivalent for the past 6 months and systemic corticosteroid use (≤20 mg·day−1 prednisone equivalent) were allowed. Furthermore, their Asthma Control Questionnaire (ACQ) score was >1.5 for 2 weeks and they were nonsmokers for ≥1 year (former smoker ≤15 pack-years).
Culturing PBECs and stimulation
PBECs were obtained by brush biopsy during bronchoscopy (P0) from healthy individuals and asthmatics, plated on six-well plates precoated with PureCol (Advanced Biomatrix, San Diego, CA, USA) and grown until confluence in BEBM medium (Lonza, Basel, Switzerland) supplemented with growth factors (Lonza) and Ciproxin (Sigma, Reading, UK) at 2 μg·mL−1. The cells were then passaged into 24-well plates (P1) in equal amounts and when confluent stimulated with recombinant human (rh)TNF-α (5 ng·mL−1) (R&D Systems, Minneapolis, MN, USA), rhIL-17A (100 ng·mL−1) (R&D Systems), rhTNF-α plus rhIL-17A (5 ng·mL−1 and 100 ng·mL−1, respectively) or no stimulus for 2 h or 16 h for the assessment of expressed RNA and released protein, respectively. We used the corticosteroid dexamethasone, which is a recognised reference standard for corticosteroids and is highly soluble in water. When indicated, 3.8 μM dexamethasone (Sigma) was used. The concentrations of TNF-α, IL-17A and dexamethasone were optimised in earlier studies [18]. As there was limited availability of PBECs the dexamethasone and other experiments were not performed with PBECs from all patients.
Detailed description of luminex, miRNA16 analysis and locked nucleic acid knockdown, mRNA and half-life analysis, immunohistochemistry, immunopurification and Western blot, TiAR knockdown and alveolar macrophage isolation and stimulation are provided in the supplementary material.
Results
PBECs from most mild and severe asthma patients are hyperresponsive to IL-17A and TNF-α and insensitive to corticosteroids
PBECs were obtained from 43 mild and 16 severe asthma patients and 16 healthy controls, the characteristics and clinical parameters of whom are summarised in table 1. Previously we have shown that human lung epithelial-like NCI-H292 cells (CRL 1848; American Type Culture Collection, Manassas, VA, USA) and PBECs in response to IL-17A and TNF-α displayed a synergistic production of CXCL-8 and IL-6 [18]. This was due to stabilisation of the encoding transcripts, which for NCI-H292 cells was shown to be dependent on an attenuated production of miR16 [20]. We confirmed the synergistic effect of TNF-α plus IL-17A on CXCL-8 production by PBECs from all individuals, but the synergistic effect was far more pronounced in PBECs from most asthma patients (supplementary figure E1a). In line herewith, PBECs from mild and severe asthma patients, compared to those from healthy controls, produced significantly higher levels of CXCL-8, IL-6, CXCL-10, granulocyte colony-stimulating factor (G-CSF) and macrophage migration inhibitory factor (MIF) (figure 1a–e) upon stimulation with TNF-α and IL-17A. No synergistic effect was seen on the production of vascular endothelial growth factor-A, CXCL-1, IL-1α, IL-1 receptor antagonist, CXCL-12 (data not shown). PBECs that showed a synergistic CXCL-8 production (<10 000 pg·mL−1 of CXCL-8) similar to those from most healthy controls are hereafter referred to as normoresponsive, whereas those with a higher response (≥10 000 pg·mL−1 of CXCL-8) are referred to as hyperresponsive (figure 1a). Variation in cell numbers for example and in metabolic activity for PBECs from both asthma groups and healthy individuals may underlie differences in CXCL-8 production. Hence, we plotted CXCL-8 production against the synergistic effect calculated as CXCL-8 production in response to TNF-α and IL-17A divided by CXCL-8 production to TNF-α alone plus that of CXCL-8 production to IL-17A alone, which is less dependent on these variables (supplementary figure E1b). There was a strong correlation between CXCL-8 levels and synergism for PBECs from all groups, indicative of little variability between PBECs from the various groups. It is clear from supplementary figure E1b that the normoresponsive PBECs can be separated well from the hyperresponsive PBECs by using 10 000 pg·mL−1 of CXCL-8, rather than by the synergistic factor.
Patient characteristics of mild and severe asthma patients and healthy controls

Enhanced and dexamethasone (dex)-insensitive production of inflammatory mediators in response to tumour necrosis factor (TNF)-α and interleukin (IL)-17A by primary bronchial epithelial cells (PBECs) from mild and severe asthma patients. PBECs from mild (n=43) and severe asthmatics (n=16) as compared to those from healthy individuals (n=16) produced significantly higher levels of a) CXCL-8, b) IL-6, c) granulocyte colony-stimulating factor (G-CSF), d) macrophage migration inhibitory factor (MIF) and e) CXCL-10, after stimulation with TNF-α plus IL-17A for 16 h. PBECs with relatively high CXCL-8 production were denoted hyperresponsive as opposed to normoresponsive PBECs with relative lowest CXCL-8 production. Data are presented as mean±sd. For statistical analyses two-tailed Mann–Whitney U-tests were used: *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001. The symbols for normo- and hyperresponsive CXCL-8 production were used in all graphs, showing that normo- and hyperresponsiveness extends to various mediators. f) CXCL-8, g) IL-6 and h) G-CSF production by i) normoresponsive PBECs (n=7) exposed to TNF-α and IL-17A for 16 h was significantly reduced by dexamethasone (3.8 μM), whereas ii) hyperresponsive PBECs (n=15) did not respond to dexamethasone. For statistical analyses two-tailed paired t-tests were used: *: p<0.05, ns: nonsignificant.
Hyperresponsive PBECs from healthy controls were markedly less hyperresponsive than PBECs from asthma patients (figure 1a). Most PBECs hyperresponsive for CXCL-8 were also hyperresponsive for IL-6, G-CSF, CXCL-10 and MIF (the red and blue symbols illustrating hyperresponsive CXCL-8 production were maintained in the graphs for the other cytokines). In a substudy with PBECs from five healthy subjects and four asthmatics, PBECs hyperresponsive to IL-17A and TNF-α were also hyperresponsive to IL-17A and lipopolysaccharide (LPS) (5 µg·mL−1; data not shown).
To determine sensitivity to corticosteroids, PBECs from mild and severe asthmatics were exposed to TNF-α plus IL-17A in the presence of dexamethasone (3.8 µM, titrated as the optimal concentration). With dexamethasone, normoresponsive PBECs (n=7) showed significantly reduced levels of CXCL-8, IL-6, G-CSF (figure 1f–h) as opposed to no significant changes by hyperresponsive PBECs (n=17). CXCL-8 production by both normo- and hyperresponsive PBECs exposed to TNF-α alone was significantly attenuated by dexamethasone, indicating that it is IL-17A that induces corticosteroid unresponsiveness in hyperresponsive PBECs (supplementary figure E1c). However, MIF and interferon-γ inducible protein-10 did not decrease in response to dexamethasone, not even in normoresponsive PBECs (supplementary figure E1d). Therefore, in addition to an exaggerated CXCL-8 production, hyperresponsive and normoresponsive PBECs are differentiated by dexamethasone-insensitive versus dexamethasone-sensitive CXCL-8 production, respectively.
Hyperresponsive PBECs correlate with neutrophilic inflammation and response of FEV1 to salbutamol
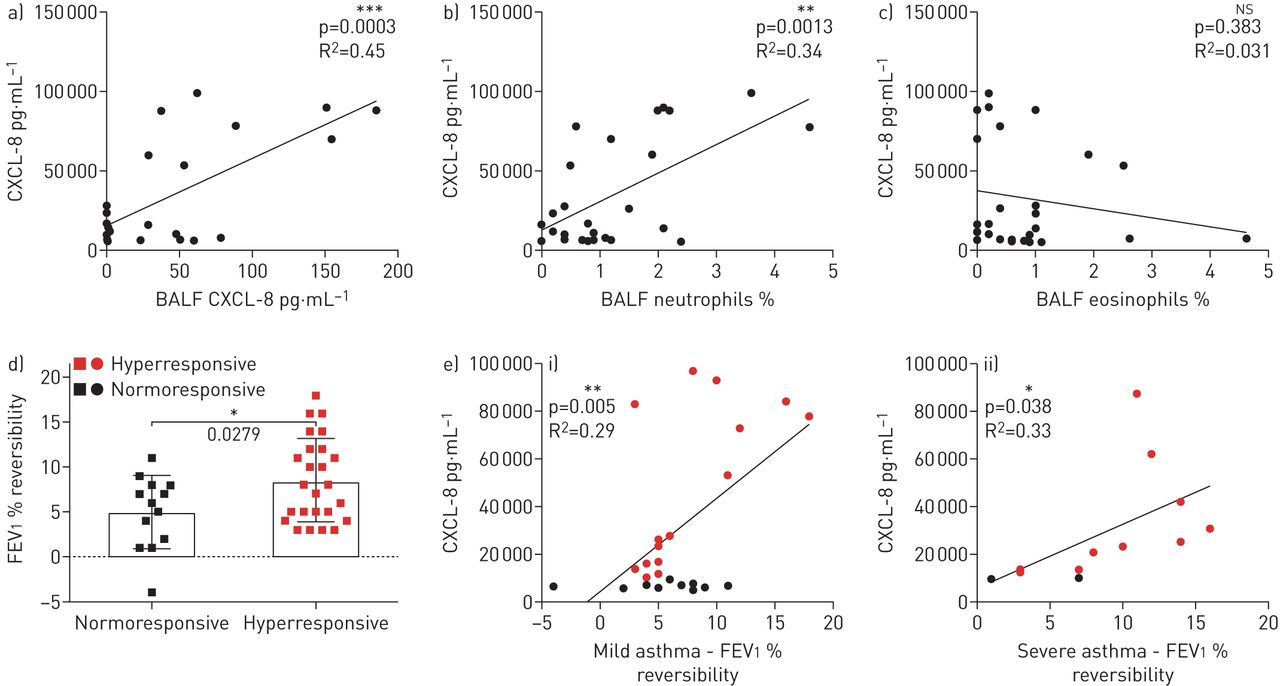
To determine whether this intrinsic feature of PBEC hyperresponsiveness may underlie key features in asthma, hyperresponsiveness was correlated to mediators in BALF and clinical parameters. As clinical and inflammatory parameters differ between mild and severe asthma, for example through the use of corticosteroids, we analysed mild and severe asthma separately. CXCL-8 production by PBECs in response to IL-17A and TNF-α significantly correlated with CXCL-8 measured in BALF of the respective mild asthma patients (figure 2a). BALF recovery (mean±sd 73±6.6%) was similar for patients, allowing for direct comparison. CXCL-8 BALF levels from healthy controls were below the lower limit of detection. Furthermore, there was a significant positive correlation with the neutrophil percentage (figure 2b) and absolute neutrophil numbers (data not shown) in BALF. No correlation was observed with eosinophil percentage (figure 2c) or with absolute eosinophil numbers.

Primary bronchial epithelial cell (PBEC) hyperresponsiveness relates to neutrophilic airway inflammation and bronchodilator-induced forced expiratory volume in 1 s (FEV1) reversibility in asthma. CXCL-8 released in vitro by PBECs from mild asthma patients (n=24) after 16 h stimulation with tumour necrosis factor (TNF)-α and interleukin (IL)-17A showed a strong positive correlation with a) CXCL-8 and b) percentage of neutrophils, but not with c) percentage of eosinophils in their corresponding bronchoalveolar lavage fluid (BALF). d) Mild and severe asthma patients (n=38) with hyperresponsive PBECs had significantly higher bronchodilator-induced FEV1 percentage reversibility compared to patients with normoresponsive PBECs. e) Bronchodilator-induced FEV1 percentage reversibility correlated positively with CXCL-8 released after 16 h stimulation with TNF-α and IL-17A, in i) mild (n=25) and ii) severe (n=13) asthma patients. Pearson's correlation test was performed with two-tailed analysis: *: p<0.05, **: p<0.01, ***: p<0.001, ns: nonsignificant.
Both baseline FEV1 % pred and bronchodilator reversibility are clinical parameters that allow subphenotyping of asthma [24, 25]. Mild and severe asthma patients with hyperresponsive PBECs had significantly higher FEV1 % reversibility compared to those with normoresponsive PBECs (figure 2d). In line herewith, hyperresponsiveness significantly correlated with the FEV1 % reversibility in mild and severe asthma patients (figure 2e). However, there was no correlation with PC20 methacholine challenge test (supplementary figure E2).
Enhanced miR16 levels in PBECs from asthma patients
We previously demonstrated that miR16 promoted CXCL8 and IL6 mRNA degradation in NCI-H292 cells [20]. miR16 levels were significantly higher in PBECs from asthma patients compared to those from healthy controls, both at baseline for unstimulated cells and after TNF-α and IL-17A exposure for 16 h (figure 3a and b, respectively). The mRNA levels of CXCL8 and IL6 in PBECs from mild and severe asthma patients were significantly higher after 2 h with TNF-α and IL-17A compared to TNF-α alone (figure 3c), indicative of a reduced mRNA degradation. In line with our previous findings [20], CXCL8 and IL6 mRNA half-lives were significantly enhanced upon exposure to TNF-α and IL-17A. However, the degradation of CXCL8 and IL6 mRNA was not different between normo- and hyperresponsive PBECs (figure 3d). Knockdown of miR16 in PBECs with a 75% transfection efficiency had no effect on CXCL8 and IL6 mRNA (figure 3e) and protein levels (figure 3f) measured after 2 h and 16 h, respectively. This indicated that CXCL8 and IL6 mRNA and protein were not modulated by miR16, contrary to what was observed earlier in NCI-H292 epithelial cells [20].

CXCL8 and IL6 mRNA half-lives are identical in normoresponsive and hyperresponsive primary bronchial epithelial cells (PBECs) despite enhanced levels of microRNA (miR)16. Relative miR16 expression was increased in PBECs a) at baseline and b) after 16 h of stimulation with tumour necrosis factor (TNF)-α and interleukin (IL)-17A in asthmatics (n=20) compared to healthy controls (n=14). Data are shown as mean±sd. For statistical analyses two-tailed Mann–Whitney U-tests were used: ***: p<0.001, **: p<0.01. c) CXCL8 mRNA and IL6 mRNA, normalised for GAPDH mRNA, were increased after 2 h of stimulation with TNF-α and IL-17A compared to TNF-α alone, both in normo- and hyperresponsive PBECs from asthma patients (n=12). d) Half-lives of CXCL8 mRNA (n=13) and IL6 mRNA (n=9) were significantly higher after 2 h with TNF-α and IL-17A stimulation compared to TNF-α alone, both in normo- and hyperresponsive PBECs from asthma patients. For statistical analyses two-tailed paired t-tests were performed: *: p<0.05, **: p<0.01, ****: p<0.0001. e) miR16 knockdown in PBECs with complementary locked nucleic acid (LNA)-miR16 and LNA-scr (scrambled control) or nontransfected (nt) as a control showed no effect on CXCL8 mRNA and IL6 mRNA 2 h after stimulation with TNF-α or TNF-α plus IL-17A, compared to not stimulated (ns) PBECs (n=6). f) As e), but for released CXCL-8 after 16 h of incubation, showing no differences with LNA-miR16 and LNA-scr or -nt after stimulation with TNF-α or TNF-α plus IL-17A, or ns (n=3). For statistical analyses two-way ANOVA and Bonferroni's multiple comparison post-test were used.
Defective cytoplasmic translocation of TiAR in hyperresponsive PBECs leads to exaggerated production of inflammatory mediators
The aforementioned results indicated that the synergistic and corticosteroid-insensitive CXCL-8 and IL-6 production by hyperresponsive PBECs to TNF-α and IL-17A was not due to attenuated mRNA degradation. An alternative explanation for the enhanced production is an altered translational control. The adenylate-uridylate (AU)-rich elements at the 3ʹ-untranslated region of mRNAs, to which AU-binding proteins and probably miR16 bind [20], are also involved in controlling translation, such as by the AU-binding protein TiAR. TiAR immunoprecipitation from the cytoplasmic fraction of normoresponsive PBECs after 2 h of TNF-α and IL-17A stimulation contained CXCL8 and IL6 mRNA, measured by quantitative PCR (figure 4a), indicating that TiAR controls translation of CXCL8 and IL6 mRNA. Normo- and hyperresponsive PBECs were immunostained for TiAR and eif3η (required for translational arrest) before and after stimulation for 2 h with TNF-α and IL-17A. TiAR co-localised with eiF3η in normoresponsive PBECs in the cytoplasm, whereas TiAR was confined to the nucleus in hyperresponsive PBECs (figure 4b). This was true for 2 h of exposure to TNF-α and IL-17A in the presence of dexamethasone (supplementary figure E3a). Identical stainings were seen after 16 h of exposure with TNF-α and IL-17A, with or without dexamethasone (data not shown). To clarify whether the failure of TiAR to translocate to cytoplasm in hyperresponsive PBECs was due to TiAR or a regulatory process facilitating TiAR translocation, hyperresponsive PBECs were exposed to arsenite, which should enforce the formation of cytoplasmic stress granules containing TiAR. Treatment with relatively low arsenite concentrations resulted in TiAR translocation and at higher concentrations led to stress granules (supplementary figure E3b). These stress granules were further confirmed by co-localisation of eiF3η with TiAR, and also were obtained in normoresponsive PBECs (supplementary figure E3c). PBECs were stained for RBP1, an endoplasmic reticulum (ER) marker, which appeared as triangles around the nucleus, whereas TiAR appeared as streaks, indicating that TiAR was not associated with ER (supplementary figure E3D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
T-cell internal antigen-1 related protein (TiAR) interacts with CXCL8 mRNA and IL6 mRNA, but fails to translocate in hyperresponsive primary bronchial epithelial cells (PBECs), leading to exaggerated production of CXCL-8. a) Western blot showing TiAR bands in cytoplasmic fraction (CyF), precleared lysate (PCL) and in the eluate, but not in the flow-through (FT). In the eluate the presence of CXCL8 and IL6 mRNA was detected by quantitative, PCR indicating that these mRNAs co-immunoprecipitated with TiAR from the cytoplasmic fraction of normoresponsive PBECs collected after 2 h of tumour necrosis factor (TNF)-α and interleukin (IL)-17A stimulation. b) Confocal images showing the co-localisation of TiAR (red) and eiF3η (green), representative of hyper- (n=8) and normoresponsive (n=4) PBECs stimulated with TNF-α and IL-17A for 2 h. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole). c) Knockdown of TiAR using three lenti-viral constructs, where the TiAR-2 construct failed to knock down TIAR (not shown). Enhanced CXCL-8 production (TNF-α plus IL-17A for 16 h) after transfection with TiAR constructs TiAR-1 and TiAR-3 in normoresponsive PBECs (n=4), whereas this did not occur in hyperresponsive PBECs (n=5). Controls are transfections with scrambled (scr) construct and nontransfected (nt) cultures. For statistical analyses, two-way ANOVA and Bonferroni's multiple comparison post-test were used. **: p<0.01; ****: p<0.0001. d) Representative Western blots showing effective knockdown of TiAR using the lenti-viral TiAR construct TiAR-3 as compared to a scrambled construct and nontransfected PBECs for both normo- and hyperresponsive PBECs. Quantified band intensities are represented in the bar chart.
To show that TiAR halts translation, we knocked down TiAR in hyperresponsive and normoresponsive PBECs. When TiAR expression was knocked down, the levels of CXCL-8 increased only in normoresponsive PBECs, whereas in hyperresponsive PBECs the knockdown had no effect on CXCL-8 production after TNF-α and IL-17A exposure (figure 4c). The knockdown of TiAR was confirmed by Western blot in both hyperresponsive and normoresponsive PBECs (figure 4d).
IL-17RA mRNA expression does not differ between normo- and hyperresponsive PBECs
In a substudy we determined whether normo- and hyperresponsive PBECs could be distinguished on the basis of differential expression of the IL-17A receptor, IL-17RA. Similar IL-17RA mRNA expression in both normo- and hyperresponsive PBECs from mild asthma patients and healthy controls was found (supplementary figure E4a). Staining of normo- and hyperresponsive PBECs on cytospins with anti-IL-17RA showed no differences in IL-17RA protein expression by immunohistochemistry, but these analyses were hampered by relatively large background staining (data not shown).
Alveolar macrophages do not display exaggerated responses to IL-17A and TNF-α
Alveolar macrophages such as bronchial epithelial cells are key players in driving immune and inflammatory responses. To determine whether alveolar macrophages showed synergistic responses to IL-17A combined with a pro-inflammatory stimulus, we purified alveolar macrophages from sputum of asthmatics and healthy controls. The macrophages and monocytes did not show any synergistic increase in CXCL-8 levels with TNF-α and IL-17A stimulations compared to TNF-α alone (supplementary figure E4b).
Discussion
Asthma is a heterogeneous disease, indicative of various mechanisms underlying the disease. Here we report a defective cytoplasmic translocation of TiAR in PBECs from asthma patients, which is paralleled by an exaggerated CXCL-8 production that is corticosteroid-insensitive when PBECs are exposed to TNF-α and IL-17A. Notably, the defect correlated with CXCL-8 levels and neutrophil numbers in BALF and with bronchodilator-induced reversibility of FEV1 of asthma patients. The extent of the defect varies between patients, in line with the heterogeneity in asthma. Of equal importance, the defect was found in PBECs from both mild and severe asthma patients. Together, this indicates that the defective TiAR translocation is a clinically relevant abnormality in asthma and underlies neutrophilic inflammation.
IL-17A in asthma has been implicated in neutrophilic and corticosteroid-insensitive responses, which may at least in part relate to the synergistic production of CXCL-8 by PBECs exposed to IL-17A and TNF-α [18]. We found that PBECs from most asthma patients showed an exaggerated synergistic effect to IL-17A and TNF-α, described here as hyperresponsiveness, contrasting with the normoresponsive PBECs from most healthy controls and some asthma patients. This hyperresponsiveness of PBECs, which is paralleled by corticosteroid insensitivity, is intriguing for a number of reasons. First, the IL-17A and TNF-α-induced CXCL-8 release by hyperresponsive PBECs in vitro correlates with CXCL-8 levels and percentage neutrophils in BALF of mild asthmatics, which strengthens the clinical relevance of hyperresponsive PBECs. Secondly, cluster analysis in the Severe Asthma Research Program (SARP) study showed that sputum neutrophilia is associated with corticosteroid insensitivity [25], substantiating earlier findings [9]. In line herewith, our results show that IL-17A triggers corticosteroid insensitivity in hyperresponsive PBECs only, indicating that there is a difference in the signalling cascade downstream of the IL-17A receptor, but distinct from the cascade involved in mRNA stabilisation. Whether the corticosteroid-insensitive inflammation by hyperresponsive PBECs fully underlies the corticosteroid-insensitive inflammation in vivo is unknown, although the specific cytokines produced by the abundant PBECs are likely to contribute significantly to airway inflammation. Thirdly, the clinical relevance of hyperresponsive PBECs is strengthened further by its correlation with bronchodilator FEV1 % reversibility. IL-17A has been linked to bronchodilator reversibility and, in line herewith, blocking the IL-17 receptor improved ACQ scores only in the group with high FEV1 percentage reversibility [13]. A similar association between post-bronchodilator FEV1 and sputum neutrophils has been shown, but not with eosinophils [14]. Thus, these findings suggest that hyperresponsive PBECs may underlie neutrophilic inflammation in asthma. Whereas we tend to distinguish eosinophilic from neutrophilic inflammation in asthma, it is important to realise that in patients with eosinophilic inflammation (usually with ≥3% sputum eosinophils), neutrophils are also present [26]. Therefore, the TiAR translocation defect is likely to be present even in asthma patients with eosinophilic inflammation, although its contribution to asthma pathophysiology may be less than that by eosinophils.
In a recent deep-phenotyping study, asthma patients with a high IL-17A profile and neutrophilic inflammation were found to experience more exacerbations [27]. IL-1β was one of the genes abundantly overexpressed in these patients. Previously we described that TNF-α combined with IL-17A can be substituted for by IL-1β, which gave similar exaggerated CXCL-8 responses by PBECs [17]. In another recent murine study IL-17A was found to synergise with IL-33 in corticosteroid-insensitive neutrophilic inflammation and enhanced airway hyperresponsiveness [28]. Together this indicates that similar analyses should be performed with IL-1β and IL-33, which may reveal a role for hyperresponsive PBECs in exacerbations. Rodhe et al. [29] showed enhanced neutrophilic inflammation upon a rhinovirus-induced exacerbation. More recently, Toussaint et al. [30] showed that neutrophil extracellular DNA traps were formed during virus-induced exacerbations, similar to that found for respiratory syncytial virus [31]. These traps are potentially antiviral as they do bind viruses [31], but at the same time also can cause airway obstruction.
Previously, we have shown that IL-17A attenuates CXCL8 mRNA degradation by reducing miR16 levels, which leads to an exaggerated CXCL-8 production. In the present study we confirmed the attenuated CXCL8 mRNA degradation upon exposure to IL-17A. However, contrary to our expectations, IL-17A-induced changes in the half-lives of CXCL8 and IL6 mRNA were similar to that in normoresponsive PBECs, whereas miR16 levels were markedly higher in hyperresponsive PBECs. These findings led us to hypothesise that the exaggerated production of CXCL-8 was due to an aberrant translational control, which made us focus on TiAR. TiAR can bind to CXCL8 mRNA and thus its failure to translocate to the cytoplasm in hyperresponsive PBECs suggests that TiAR does not halt translation of CXCL8 mRNA. Similar TiAR-binding sites are present in mRNAs encoding other cytokines, which may explain similar exaggerated production of the other cytokines. We consider it unlikely that the hyperresponsiveness of PBECs is related to differential IL-17RA expression. IL-17RA mRNA expression was similar for hyperresponsive and normoresponsive PBECs. Although the IL-17RA stainings were less conclusive, hyperresponsive and normoresponsive PBECs did not show different responses to IL-17A alone. On top of that, IL-17A equally stabilised CXCL8 and IL6 mRNA in both normoresponsive and hyperresponsive PBECs, indicating that at least part of the IL-17A downstream signalling is not different between hyper- and normoresponsive PBECs. Macrophages do not show this synergistic response to IL-17A and TNF-α, making the existence of hyperresponsive macrophages less likely. However, we cannot exclude this yet for fibroblasts, which do show a synergistic response to IL-17A and TNF-α [18].
In this study, patients and controls were well characterised, the procedures were well standardised in the subsequent studies and we have gone to great lengths to characterise the associated molecular defect, but there are a couple of limitations to this study. Although all brushes and PBECs were treated identically and there were no obvious differences in cell growth, we cannot exclude the possibility of differential growth between hyper- and normoresponsive PBECs. However, hyperresponsive PBECs, based upon CXCL-8 release by TNF-α and IL-17A were also hyperresponsive when expressed relative to TNF-α and IL-17A alone, i.e. the synergistic effect. Therefore, we conclude that the differences obtained in responsiveness to IL-17A are genuine. In severe asthma patients there were some bronchial brushes that could not be successfully cultured, so we cannot exclude a minor selection bias. The use of oral and inhaled corticosteroids by severe asthmatics on a regular basis might have influenced the mediators released by PBECs. However, PBECs were cultured a couple of weeks in vitro before stimulation with IL-17A and TNF-α, which suggests that hyperresponsiveness is an intrinsic defect. The use of corticosteroids in severe asthmatics may have affected lung function. Nevertheless, we found a correlation between hyperresponsive PBECs and FEV1 reversibility for severe asthma patients as well. There was no correlation observed between pre-bronchodilator FEV1 % pred and hyperresponsiveness in PBECs from both mild and severe asthma patients nor with percentage BALF neutrophils (data not shown). Therefore, we cannot confirm the findings by Shaw et al. [14] for which there may be various explanations. It is most likely that the number of severe asthma patients used in this correlation analysis is very low compared to their study.
For the current studies PBECs were grown submerged as opposed to the more differentiated air–liquid interface (ALI) cultures. This was done to limit the time span between sampling and testing as we appeared to lose the defect with more passages of the PBECs (data not shown). Experiments with submerged cultures required ∼10 days, whereas ALI cultures would have taken ∼28 days. Furthermore, at least in our hands, the success rate of submerged cultures was higher than for ALI cultures. And finally, adding stimuli and additional compounds to ALI cultures, with apical and basolateral domains may have complicated these quite complex studies further. As PBECs from both healthy and asthmatic individuals were treated in exact the same manner, the current differential findings with the submerged cultures are genuine. The relevance of which is clear from the correlation of hyperresponsiveness with in vivo parameters (BALF CXCL-8 and neutrophils, and FEV1) from the individuals from whom PBECs were obtained.
In summary, PBECs in the majority of mild and severe asthma patients are hyperresponsive to IL-17A and pro-inflammatory stimuli such as TNF-α and LPS, reflected by an enhanced production of CXCL-8 and other inflammatory mediators, and corticosteroid insensitivity. A defective cytoplasmic translocation of TiAR underlies this hyperresponsiveness and relates to neutrophilic inflammation and bronchodilator-induced FEV1 reversibility. Unravelling the underlying mechanisms would facilitate the development of therapeutic interventions that could reduce the burden of corticosteroid-insensitive neutrophilic inflammation in asthma.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00547-2019.Supplement
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00547-2019.Shareable
Acknowledgements
The authors would like to acknowledge Veronique Kruys and Cyril Gueydan (IBMM/CMMI, Université Libre de Bruxelles, Brussels, Belgium) for their expert opinion on TiAR and providing TiAR constructs for knockdown. We sincerely thank Tamara Dekker and Barbara Dierdorp (Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands) for luminex analyses. Our sincere thanks to Christof J. Majoor for performing bronchoscopy, Julia d'Hooghe and Annika W.M. Goorsenberg (Amsterdam UMC, University of Amsterdam) for helping with obtaining clinical data from TASMA study. The authors thank Marianne van de Pol, Suzanne Bal and Yanaika S. Sabogal Piñeros (Amsterdam UMC, University of Amsterdam) for their contributions in carrying out the RESOLVE and MATERIAL studies, respectively. The authors are very grateful to all patients and healthy controls for their participation in the studies; without their commitment and suggestions this study would not have been possible.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: A. Ravi conducted experiments, analysed data and prepared the manuscript. S. Chowdhury conducted experiments and analysed data. A. Dijkhuis conducted experiments. P.I. Bonta performed bronchoscopies, established the TASMA study and reviewed the manuscript. P.J. Sterk contributed to study design and reviewed the manuscript. R. Lutter devised the study, contributed to the experimental set-up definition and prepared the manuscript. All authors approved the final version of the manuscript.
Conflict of interest: S. Chowdhury has nothing to disclose.
Conflict of interest: A. Dijkhuis has nothing to disclose.
Conflict of interest: P.I. Bonta has nothing to disclose.
Conflict of interest: P.J. Sterk has nothing to disclose.
Conflict of interest: R. Lutter has nothing to disclose.
Conflict of interest: A. Ravi has nothing to disclose.
Support statement: This study was supported by the Netherlands Asthma Foundation (currently Lung Foundation; projects: 3.2.10.069, 3.2.07.012 and 3.2.06.031) and GSK (CRT 114696). TASMA study: this study is funded by the Netherlands Lung Foundation (grant number: 5.2.13.064JO) and The Netherlands Organization for Health Research and Development (ZonMw grant number: 90713477). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received August 3, 2018.
- Accepted May 10, 2019.
- Copyright ©ERS 2019