



Plasma mitochondrial DNA is associated with extrapulmonary sarcoidosis
- Changwan Ryu1,
- Caitlin Brandsdorfer1,
- Taylor Adams1,
- Buqu Hu1,
- Dylan W. Kelleher1,
- Madeleine Yaggi1,
- Edward P. Manning1,
- Anjali Walia1,
- Benjamin Reeves1,
- Hongyi Pan1,
- Julia Winkler1,
- Maksym Minasyan1,
- Charles S. Dela Cruz1,
- Naftali Kaminski1,
- Mridu Gulati1,2 and
- Erica L. Herzog1,2⇑
- 1Section of Pulmonary, Critical Care and Sleep Medicine, Yale School of Medicine, New Haven, CT, USA
- 2Equal contribution
- Erica L. Herzog, Section of Pulmonary, Critical Care and Sleep Medicine, Yale University, PO Box 208057, New Haven, CT, USA. E-mail: erica.herzog{at}yale.edu
Abstract
Sarcoidosis is an unpredictable granulomatous disease in which African Americans disproportionately experience aggressive phenotypes. Mitochondrial DNA (mtDNA) released by cells in response to various stressors contributes to tissue remodelling and inflammation. While extracellular mtDNA has emerged as a biomarker in multiple diseases, its relevance to sarcoidosis remains unknown. We aimed to define an association between extracellular mtDNA and clinical features of sarcoidosis.
Extracellular mtDNA concentrations were measured using quantitative PCR for the human MT-ATP6 gene in bronchoalveolar (BAL) and plasma samples from healthy controls and patients with sarcoidosis from The Yale Lung Repository; associations between MT-ATP6 concentrations and Scadding stage, extrapulmonary disease and demographics were sought. Results were validated in the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis cohort.
Relative to controls, MT-ATP6 concentrations in sarcoidosis subjects were robustly elevated in the BAL fluid and plasma, particularly in the plasma of patients with extrapulmonary disease. Relative to Caucasians, African Americans displayed excessive MT-ATP6 concentrations in the BAL fluid and plasma, for which the latter compartment correlated with significantly higher odds of extrapulmonary disease.
Enrichments in extracellular mtDNA in sarcoidosis are associated with extrapulmonary disease and African American descent. Further study into the mechanistic basis of these clinical findings may lead to novel pathophysiologic and therapeutic insights.
Abstract
Extrapulmonary sarcoidosis is a devastating disease phenotype that disproportionately affects African Americans. Enrichments in plasma mitochondrial DNA are seen in sarcoidosis and are associated with extrapulmonary disease and African American descent. http://bit.ly/2MHjAm6
Introduction
Sarcoidosis is a granulomatous disease of unknown aetiology with an unpredictable clinical course in which some patients experience self-limited or stable disease and others develop progressive, debilitating impairment with multi-organ involvement [1]. For unknown reasons, African American patients experience significant morbidity and mortality from increased rates of fibrotic lung disease and extrapulmonary manifestations [2]. Identification of mechanistic biomarkers predicting the development of fibrotic and/or extrapulmonary disease represents an unmet need because, presently, there are no accepted biomarkers for identifying patients at-risk for these aggressive disease phenotypes [3, 4].
Although it is widely accepted that granuloma formation involves an adaptive immune response [1], the pathobiological contribution of innate immunity remains less defined [5]. While studies show that alterations in macrophage proliferation [6] and activation [7] mediate granuloma formation, and differential expression of innate immune receptors [8], particularly toll like receptor 9 (TLR9) [9], demonstrates diagnostic properties in sarcoidosis [10], the mechanisms through which innate immune processes are involved remain unknown. Innate immunity is activated by receptor-mediated recognition of agonists such as pathogen-associated molecular patterns, which arise from infectious agents, and danger-associated molecular patterns, which are generated by injured cells [11]. Most sources agree that sarcoidosis results from the host response to infectious or environmental exposures [12, 13], but the innate immune response to endogenous ligands is unknown. One potential innate immune ligand is the unmethylated, CpG-rich mitochondrial DNA (mtDNA) that functions as an endogenous TLR9 agonist [14–16]. Released either non-specifically by necrotic cells [17] with the nuclear DNA-binding protein high mobility group box 1 (HMGB1) [18, 19] or via regulated processes by stressed but viable cells [14, 20], extracellular mtDNA mediates both antimicrobial and pro-inflammatory responses [21]. Experimental exposure to mtDNA or synthetic analogues activates macrophages [22] and TLR9 [10], but little is known regarding the association between mtDNA and granulomatous processes. Thus, elucidation of mtDNA's relevance in sarcoidosis, particularly regarding severe disease phenotypes, may provide mechanistic insight.
Disparate rates of fibrotic and extrapulmonary disease between Caucasian and African American sarcoidosis patients remain poorly understood [1, 23]. Epidemiological studies indicate that socioeconomic status and environmental factors do not fully account for these observations [24], and genome-wide association studies have demonstrated an increased incidence of fibroproliferative disorders among African Americans, including a subgroup of sarcoidosis [24, 25]. However, correlating genetic variants with specific, clinically relevant disease phenotypes requires further study [4]. Thus, identifying biomarkers reflective of the sarcoidosis disease state might enhance our understanding of the biological basis behind the worsened clinical outcomes observed among African Americans.
While the diagnostic and prognostic significance of extracellular mtDNA has been demonstrated in various diseases [14, 26, 27], a relationship with sarcoidosis is unknown. In this study, we used bronchoalveolar (BAL) and plasma samples from subjects obtained from two independent sarcoidosis cohorts to define an association between extracellular mtDNA and severe clinical phenotypes in this enigmatic disease.
Materials and methods
Subjects
For the discovery cohort, BAL and plasma specimens and corresponding clinical data were obtained from The Yale Lung Repository housed within the Interstitial Lung Disease Center of Excellence at Yale School of Medicine. For the validation cohort, BAL and plasma specimens and corresponding clinical data were obtained from the Genomic Information Center (GIC) of the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study. The study rationale and procedures have been previously described [28]. For the control group, biospecimens from healthy subjects without known inflammatory or lung disease were obtained from The Yale Lung Repository [26].
All human studies were performed with informed consent using protocols approved by the Institutional Review Board at each participating institution and by the GRADS GIC. Sarcoidosis diagnosis was based on current consensus guidelines [28, 29]. Clinical data included the following: disease duration; pulmonary function testing results for per cent predicted forced vital capacity (FVC % pred), forced expiratory volume after 1 s (FEV1 % pred) and diffusing capacity of the lung for carbon monoxide (DLCO % pred), and FEV1/FVC; Scadding stage; the presence of extrapulmonary disease; active or recent use of systemic therapy; and the patient-centred outcome of fatigue, as determined by the Fatigue Assessment Scale (FAS) [30].
Mitochondrial DNA quantification
Isolation and quantification of mtDNA from biospecimens were performed [14, 26] as outlined in the supplementary material.
TLR9 detection
Commercially available human TLR9-expressing HEK 293 cells (Invivogen, San Diego, CA, USA) were cultured and assayed for TLR9 activation [14] as outlined in the supplementary material.
HMGB1 quantification
Quantification of plasma HMGB1 concentrations was performed using a commercially available ELISA (Aviva Systems Biology Corp., San Diego, CA, USA) [31] as outlined in the supplementary material.
Statistical analysis
Data distribution was assessed using the D'Agostino–Pearson omnibus test. Categorical data were analysed with Fischer's exact test. Parametric comparisons were made using a t-test, and non-parametric data were compared using Mann–Whitney. Multivariate analysis was completed with multiple linear regression. Receiver operator curve (ROC) analysis was performed to determine a threshold MT-ATP6 value for extrapulmonary disease. Logistic regression models were developed to determine odds ratios. These evaluations were performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA), MedCalc (MedCalc Software, Ostend, Belgium) or SAS 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Patient population
We analysed BAL and plasma specimens from control and sarcoidosis subjects from Yale, and then we validated our findings with GRADS subjects [28]. Demographic and clinical characteristics are shown in table 1. For the derivation cohort, control and sarcoidosis subjects were recruited from the Greater New Haven area, where controls were demographically matched by age, sex, and race. For the validation cohort, we leveraged resources from the GRADS study. Notably, although Yale was a GRADS site, for our purposes we excluded the Yale GRADS subjects because they had been evaluated previously. Interestingly, Yale sarcoidosis subjects and GRADS subjects had significantly higher rates of smoking than controls. However, there was no association found between smoking and mtDNA concentrations (supplementary figure S1).
Baseline characteristics of control and sarcoidosis subjects
Extracellular mtDNA is elevated in the BAL fluid and plasma of sarcoidosis subjects in the discovery cohort
Elevated extracellular mtDNA concentrations present in the BAL fluid and plasma of various diseases lend clinically relevant insight [32]. To determine if similar findings are seen in sarcoidosis, we began by evaluating BAL and plasma samples from the relatively small Yale sarcoidosis cohort and demographically matched controls using a well-validated method that measures copy numbers of the mtDNA specific gene, MT-ATP6 [14, 26]. Relative to control subjects, we found significant increases in both the BAL fluid (3.805 versus 4.716 log copies·μL−1, p=0.008, figure 1a) and plasma (3.174 versus 5.032 log copies·μL−1, p<0.0001, figure 1b) of sarcoidosis subjects, independent of age, sex, African American race, smoking status and treatment status. Importantly, in this cohort, mtDNA concentrations were not associated with platelet counts, a common confounder of mtDNA assays [33] (Spearman r= −0.017, p=0.793, supplementary figure S2a), nor with leukocyte counts (Spearman r=0.047, p=0.818, supplementary figure S2b). While there was no correlation between MT-ATP6 copy numbers in the BAL and plasma of matched samples (Spearman r=0.344, p=0.149, figure 1c), median MT-ATP6 concentrations were an order of magnitude lower in the BAL fluid than their respective plasma sample (4.716 versus 5.060 log copies·μL−1, p=0.029, figure 1d). These data show that local and circulating concentrations of mtDNA are enriched in sarcoidosis, suggesting a connection to the disease state.

Extracellular mitochondrial DNA (mtDNA) was elevated in the bronchiolar lavage (BAL) and plasma samples of subjects in the Yale sarcoidosis cohort. Subjects with sarcoidosis in the Yale cohort displayed significantly elevated concentrations of MT-ATP6 relative to normal controls (BAL: n=9; plasma: n=50) in the a) BAL fluid (n=19) and b) plasma (n=27). c) While there was no correlation in MT-ATP6 concentrations between matched BAL and plasma samples (n=19, Spearman r=0.344, p=0.149), d) median MT-ATP6 concentrations were an order of magnitude lower in the BAL fluid than their respective plasma sample. Data are presented as log base 10 of the raw values of MT-ATP6 copies per µL of BAL fluid or plasma with median value and interquartile range.
Extracellular mtDNA is elevated in the BAL fluid and plasma of sarcoidosis subjects in the validation cohort
Sarcoidosis is a heterogeneous disease that displays differences in clinical phenotypes that vary somewhat by geographic region [34]. Thus, it was necessary to validate our findings in a larger, more heterogeneous cohort; therefore, we employed the National Institutes of Health-sponsored GRADS study, which enrolled and characterised subjects with various forms of sarcoidosis across nine US institutions from 2013 to 2015 [28]. When we repeated the above studies in the GRADS samples, we found similar increases in the BAL (3.805 versus 4.178 log copies·μL−1, p=0.022, figure 2a) and plasma (3.174 versus 5.343 log copies·μL−1, p<0.0001, figure 2b) that, as for the Yale subjects, were independent of age, sex, African American race, smoking status, and treatment status. As with the derivation cohort, there was no correlation between MT-ATP6 copy numbers in the BAL fluid and plasma (Spearman r=0.010, p=0.888, figure 2c), and median MT-ATP6 concentrations were similarly lower in the BAL fluid than plasma (4.178 versus 5.331 log copies·μL−1, p<0.0001, figure 2d). To understand the functional relevance of mtDNA in the circulation, we then stimulated the above TLR9-expressing HEK 293 cells with control and GRADS plasma. Relative to plasma from healthy controls, plasma obtained from GRADS subjects robustly resulted in TLR9 activation (0.193 versus 0.409 absorbance at 640 nm, p<0.0001, figure 3a) that significantly correlated with plasma MT-ATP6 concentrations (Spearman r=0.410, p<0.0001, figure 3b), suggesting the presence of a TLR9 ligand, specifically mtDNA, in the plasma of sarcoidosis subjects. Moreover, among GRADS subjects, plasma mtDNA copy numbers did not correlate with their respective plasma concentration of the DNA-binding protein HMGB1 (Spearman r=0.138, p=0.016, figure 3c), indicating an active process by which mtDNA is released into the circulation. These data confirm that the TLR9 agonist mtDNA is elevated in the lungs and blood of sarcoidosis subjects.

Elevations in extracellular mitochondrial DNA (mtDNA) in the bronchiolar lavage (BAL) and plasma samples of subjects with sarcoidosis were validated in the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) cohort. Subjects with sarcoidosis in the GRADS cohort displayed elevated concentrations of MT-ATP6 relative to normal controls (BAL: n=9; plasma: n=50) in the a) BAL fluid (n=205) and b) plasma (n=304). As seen with the Yale cohort, c) there was no correlation in MT-ATP6 concentrations between matched BAL and plasma samples (n=205, Spearman r=0.010, p=0.888), and d) median MT-ATP6 concentrations were an order of magnitude lower in the BAL fluid than their respective plasma sample in the GRADS cohort. Data are presented as log base 10 of the raw values of MT-ATP6 copies per µL of BAL fluid or plasma with median value and interquartile range.
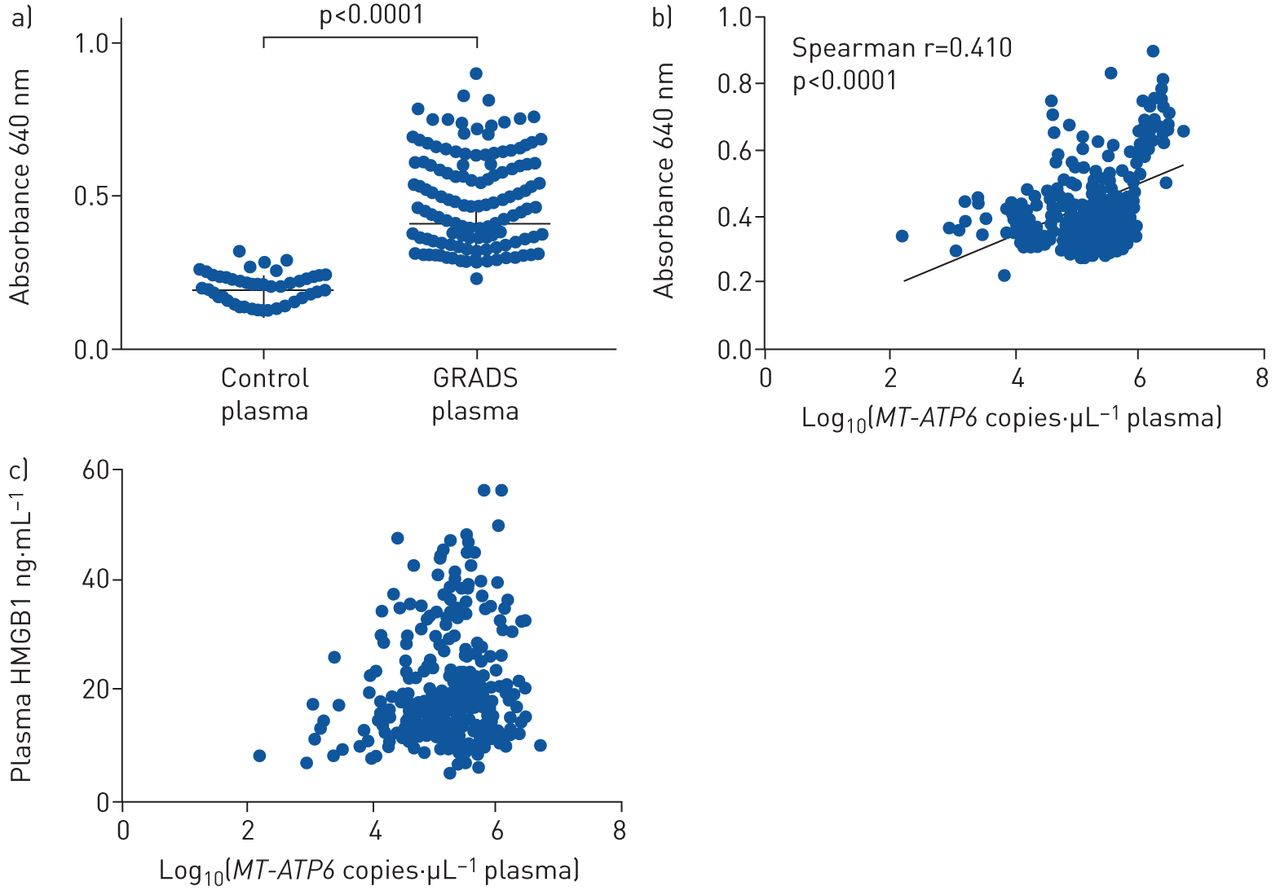
Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) plasma resulted in toll like receptor 9 (TLR9) activation that correlated with plasma mitochondrial DNA (mtDNA) concentrations. a) Plasma from GRADS subjects (n=304) resulted in substantial TLR9 activation relative to plasma from normal controls (n=50), which b) significantly correlated with plasma MT-ATP6 concentrations (log base 10 of the raw values of MT-ATP6 copies per µL of plasma). TLR9 activation is presented as median absorbance at 640 nm with interquartile range. c) Plasma MT-ATP6 copy numbers did not correlate with their respective plasma concentration of the DNA-binding protein high mobility group box 1 (HMGB1) (Spearman r=0.138, p=0.016).
Extracellular mtDNA is not increased in pulmonary sarcoidosis
Having detected substantial increases in local and circulating mtDNA in two sarcoidosis cohorts, we next sought an association with clinical phenotypes. Management strategies differentiate patients with and without lung involvement based on Scadding stage, which, despite its limitations, remains the clinical standard [35]. To determine whether mtDNA levels in the BAL fluid or plasma are elevated in subjects with known lung involvement, we stratified the Yale cohort into those lacking detectable lung involvement (stage 0/I) versus those with clear lung involvement (stages II, III and IV). Somewhat surprisingly, this approach failed to discern significant differences in the BAL (4.385 versus 4.812 log copies·μL−1, p=0.100, supplementary figure S3a) or plasma (5.038 versus 5.208 log copies·μL−1, p=0.537, supplementary figure S3b), a finding that was also seen in the GRADS BAL fluid (4.134 versus 4.169 log copies·μL−1, p=0.775, supplementary figure S3c) and plasma (5.348 versus 5.327 log copies·μL−1, p=0.119, supplementary figure S3d). When stage I subjects were omitted from the analysis, there were no significant differences between subjects with stage 0 disease and those with stage II, III or IV disease in the BAL fluid (5.127 versus 5.321 log copies·μL−1, p=0.588) or plasma (5.208 versus 5.327 log copies·μL−1, p=0.865) of the GRADS cohort, although this approach did trend towards statistical significance. Moreover, neither BAL fluid nor plasma MT-ATP6 concentrations showed any correlation with commonly used measures of lung function in either cohort, including FEV1 % pred (supplementary figure S4), FVC % pred (supplementary figure S5) and DLCO % pred (supplementary figure S6). These data show that extracellular mtDNA is increased in the BAL fluid and plasma of sarcoidosis subjects in a manner that is independent of pulmonary involvement, indicating a potential relationship with other disease features.
Plasma mtDNA is elevated in extrapulmonary disease
A clinically significant indicator of disease severity is the presence of extrapulmonary involvement, which portends significant morbidity and mortality [36, 37]. The diagnosis of organ involvement followed modified ACCESS criteria (A Case Control Etiologic Study of Sarcoidosis) as per the GRADS protocol [28], and the extrapulmonary organ systems involved are shown for Yale (supplementary table S1) and GRADS (supplementary table S2), for which dermatological, cardiac and joint involvement were the three most commonly reported extrapulmonary manifestations in both cohorts. To determine whether mtDNA was associated with this complication, BAL and plasma specimens from the Yale subjects without and with extrapulmonary disease were analysed. As shown in figure 4a, b, MT-ATP6 concentrations in the BAL fluid were similar in both cohorts regardless of the absence or presence of extrapulmonary disease (Yale: 4.730 versus 4.692 log copies·μL−1, p=0.773; GRADS: 4.064 versus 4.160 log copies·μL−1, p=0.279). However, when comparing the plasma of Yale subjects with lung-restricted disease to that of subjects with extrapulmonary involvement, MT-ATP6 concentrations were substantially elevated (4.522 versus 5.066 log copies·μL−1, p=0.001, figure 4c). These findings were repeated in the GRADS cohort (5.152 versus 5.377 log copies·μL−1, p=0.019, figure 4d). Not surprisingly, in a functional investigation of this mitochondrial-related danger-associated molecular pattern, relative to the plasma from GRADS subjects lacking extrapulmonary involvement, plasma from those with extrapulmonary disease exhibited greater TLR9 activation (0.375 versus 0.426 absorbance at 640 nm, p=0.001, figure 5a). Furthermore, these observations did not appear to be related to necrosis given that plasma MT-ATP6 copy numbers did not correlate with plasma HMGB1 concentrations among subjects with extrapulmonary disease (Spearman r=0.130, p=0.058, figure 5b). Interestingly, no organ-specific associations with plasma MT-ATP6 concentrations were found in either cohort when evaluating those without and with dermatological (supplementary figure S7a, b), cardiac (supplementary figure S7c, d) or joint disease (supplementary figure S7e, f). These findings demonstrate an association between plasma mtDNA concentrations and extrapulmonary disease.
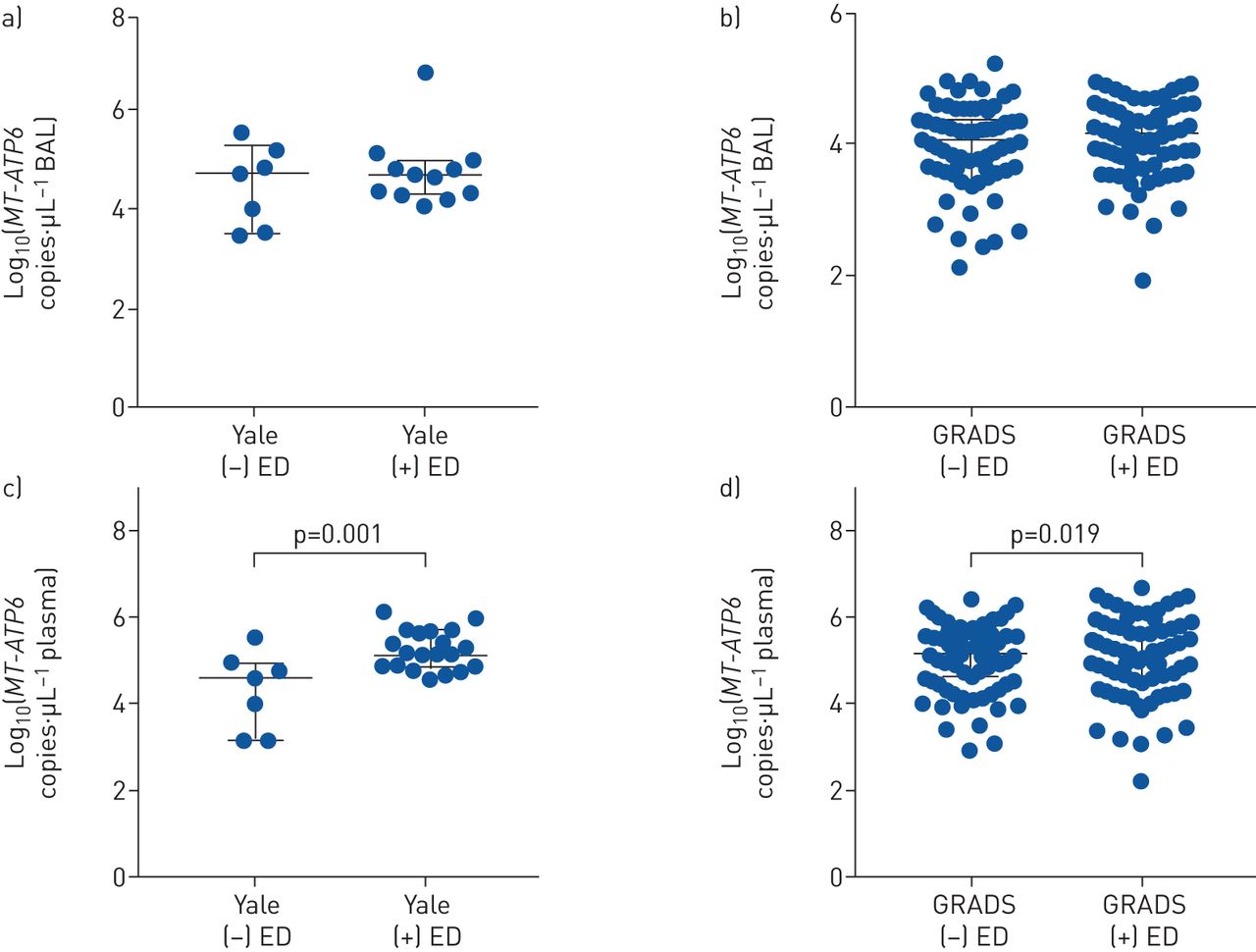
Extracellular mitochondrial DNA (mtDNA) was elevated in the plasma of sarcoidosis subjects with extrapulmonary disease (ED) in both cohorts. Median MT-ATP6 concentrations were similar in the bronchiolar lavage fluid in subjects with and without ED in both the a) Yale (n=7 versus 12, p=0.773) and b) Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) (n=83 versus 121, p=0.279) cohorts. c) However, in the Yale cohort, median plasma concentrations of MT-ATP6 were significantly elevated in subjects with ED (n=20) relative to subjects with disease limited to the lung (n=7). d) Similar results were seen in the GRADS cohort (n=90 versus 214). Data are presented as log base 10 of the raw values of MT-ATP6 copies per µL of plasma with median value and interquartile range.

Elevated plasma mitochondrial DNA (mtDNA) was associated with high odds of extrapulmonary disease (ED) and excessive fatigue. a) Plasma from Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) subjects with ED (n=214) exhibited greater toll like receptor 9 (TLR9) activation than plasma obtained from subjects lacking extrapulmonary involvement (n=90). Data presented as median absorbance at 640 nm with interquartile range. b) These observations did not appear to be related to necrosis because plasma MT-ATP6 concentrations (log base 10 of the raw values of MT-ATP6 copies per µL of plasma) did not correlate with plasma high mobility group box 1 (HMGB1) concentrations among these subjects with ED (Spearman r=0.130, p=0.058). In evaluating the clinical relevance of these findings, receiver operator curve (ROC) analysis of the Yale cohort revealed that a plasma MT-ATP6 copy number of 4.71 log copies·µL−1 reliably stratified subjects for low or high odds of ED; c) subjects whose plasma MT-ATP6 concentrations exceeded this threshold value had significantly increased odds of ED in the Yale (OR 8.500, 95% CI 1.964–36.790, p=0.004) and GRADS (OR 2.429, 95% CI 1.839–3.208, p<0.0001) cohorts. Among subjects with ED, subjects who reported elevated fatigue scores as measured by the Fatigue Assessment Scale (FAS) had significantly higher levels of plasma MT-ATP6 than participants reporting normal fatigue scores in both the d) Yale (n=5 versus 15) and e) GRADS (n=43 versus 171) cohorts. Data are presented as log base 10 of the raw values of MT-ATP6 copies per µL of plasma with median value and interquartile range.
Elevated plasma mtDNA is associated with high odds of extrapulmonary disease
We then evaluated the clinical relevance of these results given that, because patients with extrapulmonary sarcoidosis are often asymptomatic [38], an easily measured blood biomarker identifying patients at-risk for extrapulmonary disease will be of great clinical utility. ROC analysis on the Yale cohort revealed that a plasma MT-ATP6 copy number of 4.71 log copies·µL−1 can reliably stratify subjects for low odds (<4.71 log copies·µL−1) or high odds (≥4.71 log copies·µL−1) of extrapulmonary disease (area under the curve (AUC) 0.836, p=0.001, supplementary figure S8). Subjects with plasma MT-ATP6 concentrations ≥4.71 log copies·µL−1 had substantially increased odds of extrapulmonary disease at the time of evaluation (OR 8.500, 95% CI 1.964–36.790, p=0.004, figure 5c), independent of age, sex, African American race, smoking, treatment and stage IV disease. The predictive ability of this threshold value was validated in the GRADS cohort, for which, following adjustment for relevant covariates, subjects with plasma MT-ATP6 concentrations ≥4.71 log copies·µL−1 were also more likely to have extrapulmonary involvement (OR 2.021, 95% CI 1.494–2.735, p<0.0001, figure 5c). In keeping with these data, a significant association was found between plasma mtDNA and the patient-centred outcome of fatigue in both cohorts. Here, relative to participants reporting normal fatigue scores, subjects who reported elevated fatigue scores displayed significantly higher levels of plasma MT-ATP6 concentrations (Yale: 4.791 versus 5.516 log copies·μL−1, p=0.004, figure 5d; GRADS: 5.341 versus 5.534 log copies·μL−1, p=0.005, figure 5e). These findings show that elevated plasma mtDNA is a marker of both extrapulmonary disease and fatigue, perhaps reflecting the ongoing systemic inflammation that contributes to this condition.
Extracellular mtDNA is increased in the BAL fluid and plasma of African American sarcoidosis subjects
The data presented above indicate that extracellular mtDNA is a marker of extrapulmonary disease in two sarcoidosis cohorts. This led to the question of whether elevated extracellular mtDNA might also be seen in African American subjects, who, relative to their Caucasian counterparts, are at higher risk for severe disease phenotypes [2]. To this end, BAL fluid and plasma mtDNA concentrations were compared between participants of Caucasian and African American descent in both cohorts. Subject characteristics between Caucasians and African Americans are shown for Yale (supplementary table S3) and GRADS (supplementary table S4). In the Yale cohort, compared to Caucasians with sarcoidosis, BAL fluid MT-ATP6 concentrations were not increased in African Americans (4.385 versus 4.874 log copies·μL−1, p=0.221, figure 6a). However, MT-ATP6 concentrations were substantially elevated in the plasma of African Americans in the Yale (4.792 versus 5.564 log copies·μL−1, p=0.004, figure 6b) and GRADS (4.793 versus 5.010 log copies·μL−1, p=0.006, figure 6c) cohorts. These findings in the plasma were independent of age, sex, smoking, treatment, Scadding stage and extrapulmonary disease. Additionally, relative to the plasma from Caucasian GRADS subjects, African American GRADS plasma demonstrated greater TLR9 activation (0.394 versus 0.448 absorbance at 640 nm, p<0.0001, figure 7a), and plasma MT-ATP6 concentrations were independent of plasma HMGB1 concentrations (Spearman r=0.035, p=0.799, figure 7b). These data demonstrate that the TLR9 agonist mtDNA is elevated in the circulation of African American sarcoidosis subjects in two independent cohorts.
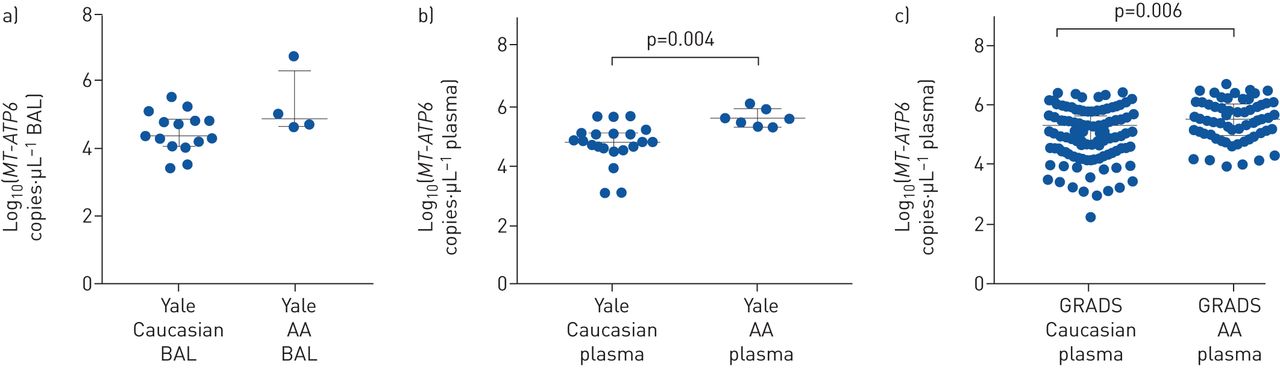
Extracellular mitochondrial DNA (mtDNA) was elevated in the plasma of African American (AA) sarcoidosis subjects. a) Median MT-ATP6 concentrations were similar in the bronchoalveolar lavage (BAL) fluid of Caucasian (n=15) and AA (n=4) sarcoidosis subjects in the Yale cohort. b) However, AA subjects (n=7) had robustly increased plasma MT-ATP6 concentrations compared with Caucasian subjects (n=20); c) these findings were subsequently validated in the much larger and more diverse Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) cohort (n=66 versus 238). Data are presented as log base 10 of the raw values of MT-ATP6 copies per µL of BAL fluid or plasma with median value and interquartile range.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Plasma mitochondrial DNA (mtDNA) provided race-specific associations with extrapulmonary disease (ED). a) Plasma from African American (AA) Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) subjects (n=54) resulted in greater toll like receptor 9 (TLR9) activation than plasma obtained from Caucasian GRADS subjects (n=160) with ED. Data presented as median absorbance at 640 nm with interquartile range. b) Plasma MT-ATP6 concentrations (log base 10 of the raw values of MT-ATP6 copies per µL of plasma) were independent of plasma high mobility group box 1 (HMGB1) concentrations (Spearman r=0.035, p=0.799). c) In subjects with ED, AA GRADS subjects had significantly elevated median concentrations of MT-ATP6 compared with Caucasian GRADS subjects. Data are presented as log base 10 of the raw values of MT-ATP6 copies per µL of plasma with median value and interquartile range. d) Forrest plot depicting odds of ED based on high plasma MT-ATP6 concentrations (≥4.71 log copies·μL−1; OR 2.021, 95% CI 1.494–2.735, p<0.0001), AA race (OR 2.497, 95% CI 1.271–4.905, p=0.008) and high plasma MT-ATP6 with AA race (OR 4.700, 95% CI 2.375–9.300, p<0.0001), which imparted the highest odds of ED. Caucasian subjects with high plasma MT-ATP6 had lower odds of ED than their AA counterparts (OR 2.293, 95% CI 1.084–4.849, p=0.030).
Extracellular mtDNA provides race-specific associations with extrapulmonary disease
After finding that plasma mtDNA is elevated in African American sarcoidosis subjects, we explored whether this provided race-specific associations with extrapulmonary disease, a frequent complication observed among African Americans [2]. Only the GRADS cohort had a sample size suitable for this analysis, and profound racial differences emerged. Relative to Caucasian race, African American race was independently associated with extrapulmonary disease (OR 2.497, 95% CI 1.271–4.905, p=0.008). This was further reflected in the plasma; when compared to Caucasians with extrapulmonary disease, African Americans with extrapulmonary disease had higher MT-ATP6 concentrations (5.348 versus 5.482 log copies·μL−1, p=0.021, figure 7c). A logistic regression model was then developed to determine the odds of extrapulmonary disease based on the previously derived threshold MT-ATP6 copy number of 4.71 log copies·μL−1 and African American race, and ROC analysis of this multivariate model revealed an AUC of 0.611. African Americans with plasma MT-ATP6 concentrations ≥4.71 log copies·μL−1 had the highest odds of having extrapulmonary disease (OR 4.700, 95% CI 2.375–9.300, p<0.0001, figure 7d). In fact, Caucasians with plasma MT-ATP6 concentrations ≥4.71 log copies·μL−1 were less likely to have extrapulmonary disease than their African American counterparts (OR 2.293, 95% CI 1.084–4.849, p=0.030, figure 7d). These findings show that elevated plasma mtDNA concentrations convey important information regarding the odds of African American sarcoidosis subjects having extrapulmonary disease.
Discussion
In a novel analysis of two independent sarcoidosis cohorts, we found a significant association between excessive extracellular mtDNA concentrations, extrapulmonary involvement and racial differences in disease. Specifically, enriched mtDNA copy numbers were found in the BAL and plasma samples of subjects with sarcoidosis, for which increases in the latter compartment were robustly associated with extrapulmonary disease. In addition, increased mtDNA concentrations were seen in African American subjects, an at-risk population for poor disease outcomes.
Since its initial report as a mediator of inflammatory joint disease, the pathogenic significance of extracellular mtDNA has been increasingly recognised [32]. The extracellular release of mtDNA occurs either non-specifically, typically in response to cellular stress or necrosis, or actively through extracellular vesicles [32]. Because we failed to detect an association between levels of MT-ATP6 and the commonly used necroptosis marker HGMB1, we believe this extracellular mtDNA accumulation results from a mechanism other than or in addition to necroptosis. Interestingly, trafficking of mtDNA via extracellular vesicles mediates cell-to-cell signal transduction as part of the inflammatory response [39], likely via activation of TLR9 [14] and STING [40] pathways. Because we found that sarcoidosis plasma showed robust TLR9 activating capacity, we believe that the most likely functional ramification of our work relates to activation of this pathway. However, alternate explanations may exist. Given that some postulate an infectious aetiology for sarcoidosis [34], it is possible that extracellular mtDNA is acting in an antimicrobial facility by activating the NLRP3 inflammasome [41], forming extracellular traps [42] or provoking mitochondrial antiviral signalling [43]. As a mediator of inflammatory, infectious and fibrosing processes, further mechanistic and functional study of mtDNA in sarcoidosis could yield novel insights into its enigmatic aetiology.
Our work also presents new observations regarding the association between mtDNA and sarcoidosis phenotypes. The finding that circulating mtDNA was elevated in subjects with extrapulmonary disease, but not pulmonary granuloma formation, may reflect previously unrecognised differences in the mechanism between the granulomatous response to inhaled versus systemically delivered antigen. It should be noted that plasma mtDNA concentrations were an order of magnitude higher than those in the BAL fluid, which may reflect true biology, technical artefact in BAL sampling and processing (such as dilutions involved in obtaining BAL fluid), or yet to be identified experimental or clinical factors. Additional work is required to determine how mtDNA, pulmonary sarcoidosis and extrapulmonary involvement are linked.
Perhaps our most exciting finding is the observed racial differences in mtDNA, especially with aggressive clinical phenotypes. Genome-wide association studies have identified susceptible genetic variants for sarcoidosis among African Americans [44], and racial differences in innate immunity have been demonstrated, particularly in TLR9 polymorphisms related to breast cancer [45] and infectious granulomas [46], and in TLR9 expression in systemic lupus erythematosus [47]. An association between TLR9 and sarcoidosis has been reported [48], although racial differences have not been explored. However, an association with race and TLR4 variants has been shown [49], and because crosstalk exists between TLR4 and TLR9 [50], it is possible that an endogenous TLR9 agonist results in excessive inflammation that then feeds into TLR4, leading to significant injury and extrapulmonary involvement. This hypothesis will require further study with overexpression and knockdown strategies.
While novel and provocative, our study has several limitations. We have not identified the tissues from which mtDNA originates, which will include identifying the cell of origin, mechanisms of release, association with mitochondrial function and regulation of extracellular trafficking. Despite these relatively minor limitations, our work provides strong evidence supporting circulating mtDNA as a biomarker of organ involvement and racially disparate clinical presentations of sarcoidosis. Further investigation may lead to new therapeutic avenues for this poorly understood and difficult to treat disease.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01762-2018.Supplement
Supplementary figure S1 ERJ-01762-2018.Figure_S1
Supplementary figure S2 ERJ-01762-2018.Figure_S2
Supplementary figure S3 ERJ-01762-2018.Figure_S3
Supplementary figure S4 ERJ-01762-2018.Figure_S4
Supplementary figure S5 ERJ-01762-2018.Figure_S5
Supplementary figure S6 ERJ-01762-2018.Figure_S6
Supplementary figure S7 ERJ-01762-2018.Figure_S7
Supplementary figure S8 ERJ-01762-2018.Figure_S8
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01762-2018.Shareable
Acknowledgements
We are extremely grateful to all our sarcoidosis patients and control subjects who generously donated their time and specimens for our studies. We thank all the GRADS investigators and GIC members for their hard work and contributions.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: C. Ryu performed experiments and statistical analysis and analysed data; C. Brandsdorfer performed experiments and analysed data; T. Adams performed experiments; B. Hu performed statistical analysis; D.W. Kelleher performed experiments; M. Yaggi performed experiments; E.P. Manning analysed data; A. Walia performed experiments; B. Reeves performed experiments; H. Pan assisted with statistical analysis; J. Winkler performed experiments; M. Minasyan analysed data; C.S. Dela Cruz analysed data; N. Kaminski analysed data; M. Gulati recruited subjects, procured biospecimens and analysed data; and E.L. Herzog conceived the experimental design and analysed data. All authors participated in manuscript preparation and provided final approval of the submitted work.
Support statement: C. Ryu was supported by grants from the Parker B. Francis Foundation and the Foundation for Sarcoidosis Research. E.P. Manning was supported by T32HL007778 grant. N. Kaminski was supported by U01HL122626, UH3HL123886 and R01HL127349 grants. E.L. Herzog was supported by R01HL109233, R01HL125850 and U01HL112702 grants, and grants from the Gabriel and Alma Elias Research Fund and the Greenfield Foundation. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: C. Ryu has nothing to disclose.
Conflict of interest: C. Brandsdorfer has nothing to disclose.
Conflict of interest: T. Adams has nothing to disclose.
Conflict of interest: B. Hu has nothing to disclose.
Conflict of interest: D.W. Kelleher has nothing to disclose.
Conflict of interest: M. Yaggi has nothing to disclose.
Conflict of interest: E.P. Manning has nothing to disclose.
Conflict of interest: A. Walia has nothing to disclose.
Conflict of interest: B. Reeves has nothing to disclose.
Conflict of interest: H. Pan has nothing to disclose.
Conflict of interest: J. Winkler has nothing to disclose.
Conflict of interest: M. Minasyan has nothing to disclose.
Conflict of interest: C.S. Dela Cruz has nothing to disclose.
Conflict of interest: N. Kaminski has nothing to disclose.
Conflict of interest: M. Gulati has nothing to disclose.
Conflict of interest: E.L. Herzog has nothing to disclose.
- Received September 18, 2019.
- Accepted May 27, 2019.
- Copyright ©ERS 2019