



When insulin has to work hard to keep the sugar at bay the upper airway collapses away
- 1Neuroscience Research Australia (NeuRA) and the School of Medical Sciences, University of New South Wales, Sydney, Australia
- 2Dept of Medicine, Bnai-Zion Medical Center and the Technion, Haifa, Israel
- Danny J. Eckert, Neuroscience Research Australia (NeuRA), PO Box 1165, Randwick, Sydney, New South Wales, Australia, 2031. E-mail: d.eckert{at}neura.edu.au
Abstract
Insulin resistance may increase upper airway collapsibility and directly contribute to sleep apnoea pathogenesis http://ow.ly/10BTf9
Obesity is a major risk factor for both obstructive sleep apnoea (OSA) and metabolic disease. As obesity rates continue to rise, so too does the prevalence of OSA and metabolic disorders. Indeed, recent community sample data from over 2000 adults aged 40–85 years in Switzerland indicate that up to 50% of men and almost a quarter of women have apnoea–hypopnoea indices (AHI) within the moderate to severe range (>15 events·h−1 sleep) [1]. Insulin resistance, a strong predictor for the development of type 2 diabetes [2], is being recognised earlier with prevalence rates in children varying between 3 and 44% [3]. Thus, OSA and insulin resistance are major health issues.
Given the shared link with obesity, the connection between metabolic disruption and OSA is not new. Indeed, OSA has been implicated in the pathogenesis of metabolic dysfunction [4–6]. Furthermore, a “bidirectional” or “reciprocal” relationship, or the possibility that OSA is a manifestation of the metabolic syndrome, has been discussed in several publications [7–10]. However, while evidence continues to grow that sleep-disordered breathing and sleep disruption can worsen metabolic function including insulin resistance [11], there is currently limited direct evidence to support the concept that early biomarkers of metabolic dysfunction such as insulin resistance predispose to OSA [12, 13]. Thus, the chicken or the egg causality dilemma remains.
The new study by Llanos et al. [14] in this issue of the European Respiratory Journal takes us one step closer to solving this dilemma. Specifically, the authors have provided elegant insight into this question by going to the source; evaluating the relationship between pre- diabetes (i.e. insulin resistance), and “pre-OSA” (i.e. individuals with vulnerable upper airways due to high mechanical load from morbid obesity but without frank OSA (AHI <10 events/h sleep)). This unique approach has allowed the investigators to study upper airway collapsibility during sleep, using the gold standard critical closing pressure or “Pcrit” technique, without the confounding effects of prior exposure to repetitive severe hypoxaemia and sleep fragmentation, both of which contribute to metabolic sequelae [5, 15]. Accordingly, consistent with insulin resistance preceding the decline in pharyngeal stability, the reported positive correlation between insulin resistance and upper airway collapsibility and the ∼4 cmH2O increase in Pcrit when the group was dichotomised according to high versus low insulin resistance cannot be explained by the presence of severe sleep-disordered breathing.
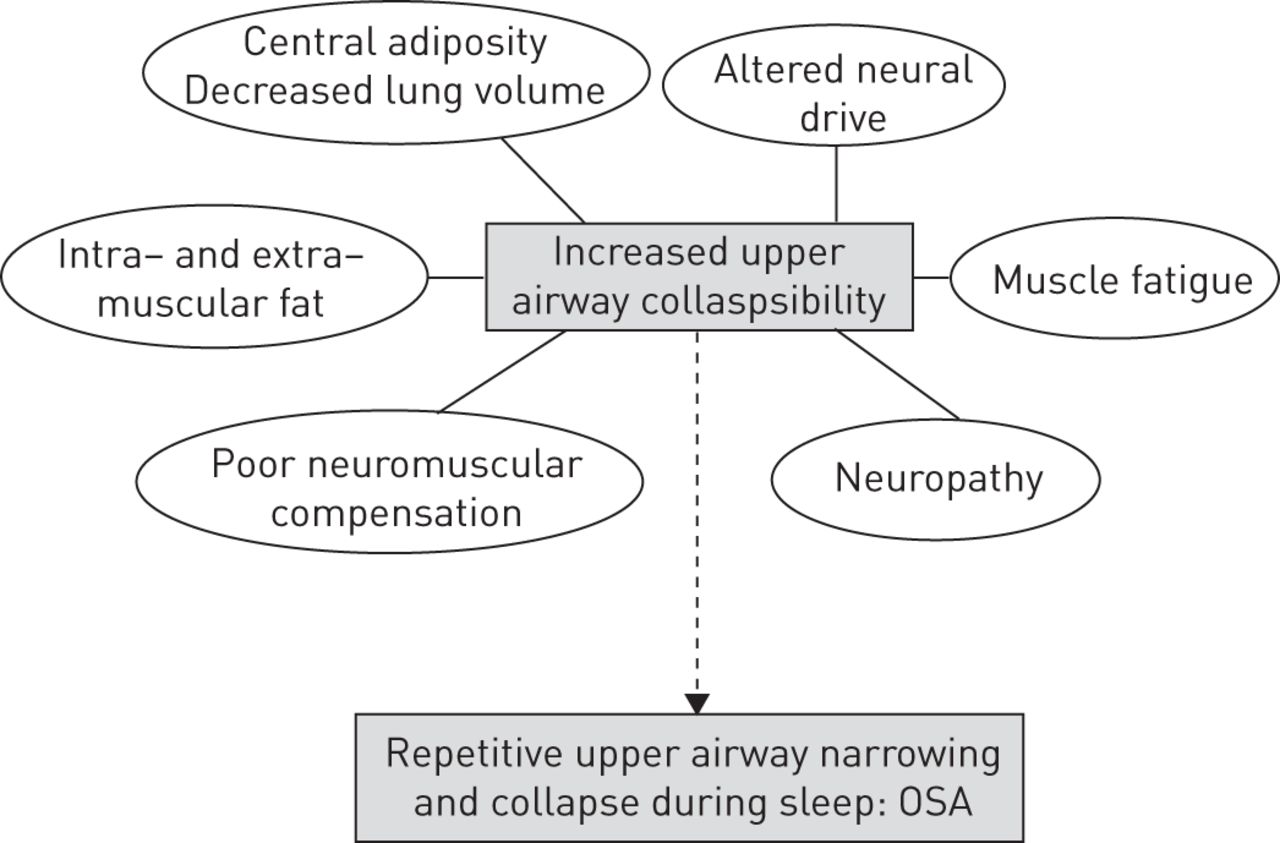
In addition to a causal role, these novel findings suggest that insulin resistance may also accelerate the development of OSA and its aggravation once manifested. Thus, the following question arises: what has the pancreas/insulin resistance got to do with upper airway collapsibility? As discussed below, there are several plausible mechanisms by which insulin resistance may increase pharyngeal collapsibility and possibly cause frank OSA, each of which will require careful future investigation (summarised in figure 1).
{kind=link}
Schematic indicating the potential mechanisms by which insulin resistance might contribute to increased upper airway collapsibility and ultimately obstructive sleep apnoea (OSA). Some of the interconnections have been omitted for clarity. Refer to the text for further detail.
Pharyngeal stability is dependent on the interaction between anatomical or static properties of the upper airway and the dynamic function of the dilator muscles [16]. Static properties include: cross sectional area, pharyngeal length and structural composition. Dynamic properties include: neural drive, coordination and muscle efficiency. The anatomic features govern the static shape and size of the upper airway whereas the dynamic components mediate the ability of the pharyngeal dilators to compensate for mechanical loads to the upper airway (e.g. obesity chronically or transient repetitive narrowing and closure as occurs during sleep in OSA). The Llanos et al. study [14] used the “passive Pcrit” technique to focus on the effects of insulin resistance on the static properties. As highlighted by the authors, factors that crowd the upper airway directly such as preferential ectopic fat accumulation in the tongue or peripharyngeal tissues with insulin resistance will increase upper airway collapsibility. Indirectly, visceral fat accumulation which is closely linked to insulin resistance will lower lung volume during sleep and make the upper airway more prone to collapse via a loss of tracheal traction [17]. To determine if these regional changes in fat distribution are indeed the key mechanisms explaining the current findings, sophisticated imaging approaches of the key upper airway structures and lateral fat pads of the neck combined with respiratory physiology work is required [18–20].
Although not the focus of the present study, insulin resistance may also alter the dynamic properties of the upper airway. The pharyngeal dilator muscles receive input from wakefulness active neurons, local reflex mechanisms and central drive (e.g. pattern generator neurons to the largest upper airway muscle, genioglossus) [21]. The passive Pcrit” technique used in the current study [14] was designed to reduce upper airway muscle activity by minimising local reflex input with continuous positive airway pressure. However, some individuals generate substantial reflex activation during these measurements [22, 23] and centrally controlled muscle activity remains, as evident by increases in passive Pcrit during rapid eye movement (REM) sleep [22] and anaesthesia [24] when compared to non-REM sleep. Thus, while there is currently no evidence to indicate that insulin resistance alters the central mechanisms controlling the residual dilator muscle activity during the conditions used for passive Pcrit measurement, the differences observed with insulin resistance in the current study may be, at least in part, due to impaired neuromuscular responses.
For example, obesity, primarily central obesity, is associated with increased circulating levels of inflammatory cytokines, some of which yield somnogenic central nervous system activity [25]. Obesity is also associated with blunted neuromuscular compensatory responses [25, 26]. It is conceivable that insulin resistance may augment these effects. Patients with untreated OSA are more prone to upper airway muscle fatigue [27, 28]. Insulin resistance is associated with lower leg muscle fatigue during a stair climbing task [29]. Thus, if insulin resistance also increases upper airway muscle fatigue, this could contribute to increased upper airway collapsibility.
An alternate mechanism by which insulin resistance might increase passive Pcrit and the predisposition or progression of OSA is via damage to the soft tissues surrounding the pharynx. Diabetic neuropathy may impair the ability of the dilator muscles to protect pharyngeal patency [30]. However, diabetic neuropathy usually develops after prolonged diabetes mellitus, and typically first manifests as a sensory peripheral neuropathy. The role of systematic sensorimotor impairment in OSA pathogenesis is also unclear [27, 31]. Thus, while this mechanism cannot be discounted, the occurrence of a relevant asymptomatic insulin resistance-induced neuropathy confined to the upper airways, either sensory or motor, would appear unlikely. Similarly, while obesity and OSA are associated with structural alteration and inflammatory reactions in the extracellular matrix of upper airway tissue [32], the role of insulin resistance in these processes has not been evaluated.
In summary, while many uncertainties regarding the underlying mechanisms remain, the study in this issue of the European Respiratory Journal by Llanos et al. [14] provides the first empiric evidence for a pathogenic contribution from insulin resistance to the primary cause of OSA, increased pharyngeal collapsibility. To extend the generalisability of this work beyond morbidly obese women (the average BMI of the participants was 48 kg·m−2), it will be important to determine if the reported relationships are also present in lean subjects, in men, and in longitudinal cohorts. Indeed, in light of recent findings [33, 34], morbidly obese individuals who do not have OSA likely have “supranormal” neuromuscular compensatory mechanisms that protect them from OSA. This concept is further supported by >50% of the study participants having a passive Pcrit above −2 cmH2O and elevated AHIs during REM sleep into the pathogenic range when neuromuscular compensatory mechanisms are reduced or absent. However, given the differences in OSA pathophysiology between the sexes, it will be challenging to extend the current design to obese men with insulin resistance who do not have OSA as these individuals are rare. It will also be of interest to determine if other contributors to OSA pathophysiology that have been implicated with insulin resistance such as heightened peripheral chemosensitivity [35] cause respiratory control instability during sleep and have a causal role in OSA pathogenesis and/or disease progression. Finally, the intriguing possibility that insulin “sensitisers” may improve upper airway collapsibility in patients with OSA and insulin resistance warrants further investigation.
Footnotes
Support statement: D.J. Eckert is supported by a National Health and Medical Research Council (NHRMC) of Australia. R.D. Wright Fellowship (1049814). Funding information for this article has been deposited with FundRef.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received March 22, 2016.
- Accepted March 23, 2016.
- Copyright ©ERS 2016