



Experimental rhinovirus 16 infection in moderate asthmatics on inhaled corticosteroids
- Peter T. Adura1,
- Eleanor Reed2,
- Jonathan Macintyre3,
- Ajerico del Rosario3,
- James Roberts2,
- Rachel Pestridge2,
- Rona Beegan2,
- Christine B. Boxall2,
- Chang Xiao2,
- Tatiana Kebadze3,
- Juliya Aniscenko3,
- Victoria Cornelius1,
- James E. Gern4,
- Phillip D. Monk2,
- Sebastian L. Johnston3 and
- Ratko Djukanović1⇑
- 1Clinical and Experimental Sciences, Southampton NIHR Respiratory Biomedical Research Unit, University of Southampton Faculty of Medicine, Southampton
- 2Synairgen Research Ltd, Southampton
- 3National Heart and Lung Institute and Centre for Respiratory Infections, Imperial College, MRC and Asthma UK Centre in Allergic Mechanisms of Asthma, Imperial College NHS Trust NIHR Comprehensive Biomedical Research Centre, London, UK
- 4University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
- R. Djukanović, Clinical and Experimental Sciences, Southampton NIHR Respiratory Biomedical Research Unit, University of Southampton Faculty of Medicine, Mailpoint 810, Level F, Sir Henry Wellcome Laboratories, South Block, Southampton General Hospital, Southampton SO16 6YD, UK. E-mail: r.djukanovic{at}soton.ac.uk
To the Editor:
The majority of asthma exacerbations are associated with respiratory virus infections, mostly rhinoviruses (RVs) [1], due to enhanced inflammation in the airways [2]. These occur despite symptom control with inhaled corticosteroids (ICS) [3]. Experimental RV infection is a valuable tool for studying virus-induced exacerbations [2, 4], but has, to date, involved only corticosteroid-naïve asthmatics. We have, therefore, modified a validated infection protocol [4] to inoculate 11 subjects whose asthma was well controlled with ICS. As this was the first experimental infection in patients at risk of severe exacerbations, a cautious study design was implemented. All subjects were followed-up twice daily by SMS text messages during the study. We used RV16, a strain used safely in previous studies, which replicates in vitro to a similar extent but induces less inflammation and cell death than other strains [5]. We also chose a 10-fold lower inoculation dose of the same stock used in previous studies [6]. The design allowed for dose escalation if necessary (this proved to be unnecessary as all subjects developed cold symptoms at this dose). As a final precaution, the delivery device generated particles of 30–100 μm, restricting delivery to the nose (aerosols ≥16 μm are deposited in the upper respiratory tract (URT) [7]), thus closely mimicking natural infection, i.e. limiting direct lung exposure during inoculation.
Symptoms of URT infection, asthma, and measurements of lower respiratory tract (LRT) function were recorded post-inoculation. Infection was confirmed by quantitative (q)PCR for RV16 in nasal lavage and sputum and by determining serum anti-RV16 titres. At least a ≥four-fold increase in titres in convalescent serum or shedding of RV16 in the airways was evidence of successful infection. We also studied innate immune responses by extracting mRNA from sputum cell pellets and whole blood cells, and by quantifying the induction of virus-response genes interferon (IFN)-β, C-X-C motif chemokine 10 (CXCL10), myxoma resistance protein A (MxA) and 2',5'-oligoadenylate synthetase (OAS). Furthermore, CXCL10 protein was measured by ELISA in nasal lavage and sputum supernatants. Finally, blood was collected before RV16 infection, treated with polyinosinic-polycytidylic acid (poly(I:C)) at a dose of 100 μg·mL−1 (Invivogen, San Diego, CA, USA) or 0.9% NaCl as control for 2 h and stored in PAXgene tubes (Qiagen, Crawley, UK) for IFN-β gene expression analysis by qPCR as a surrogate for virus-induced, Toll-like receptor 3-mediated responses [8] and to assess the value of this assay as a predictor of severity of infection.
All 11 subjects were successfully infected without any serious adverse effects, withdrawal because of safety concerns the patients, or requirement for treatment beyond extra bronchodilator use. All developed increased cold symptoms (fig. 1a), peaking at a mean (95% CI) 3.7 (2.9–4.6) days post-inoculation. ANOVA showed significant symptom differences over time (p<0.01) with a significant quadratic trend (p<0.01); mean (95% CI) of differences between baseline and peak URT symptom scores was 8.6 (7.3–9.9) (p<0.01). Asthma symptoms (fig. 1a) peaked at a mean (95% CI) 5.8 (4.0–7.6) days post-inoculation. ANOVA showed significant differences over time (p=0.01), with a significant quadratic trend (p<0.01). The mean (95% CI) of differences between baseline and observed peak values were 7.0 (4.2–9.8) (p<0.01) for asthma scores and 3.2 (1.4–4.9) (p<0.01) for bronchodilator use. The mean (95% CI) period between peak cold and peak asthma symptoms (fig. 1a) was 2.1 (0.3–3.9) days. In contrast, there was no significant reduction in lung function post-inoculation.

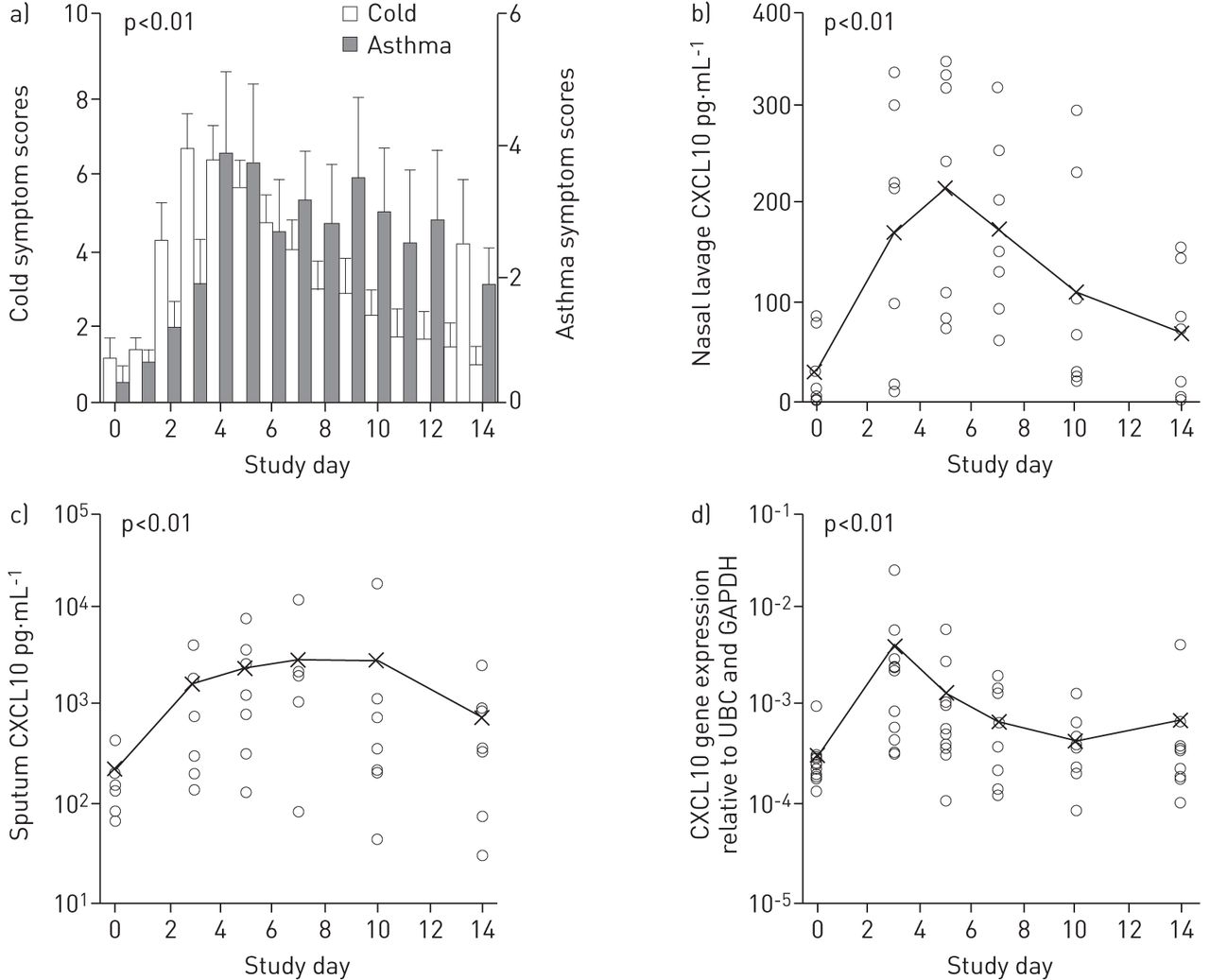{kind=link}
Key clinical and biomarker results of the study. a) Both cold and asthma symptom scores increased significantly, b) C-X-C motif chemokine 10 (CXCL10) concentrations in nasal lavage, c) CXCL10 concentrations in sputum, and d) induction of CXCL10 gene in peripheral blood cells. UBC: ubiquitin-c; GAPDH: glyceraldehyde 3-phosphate dehydrogenase. All the changes were significant (p<0.01), as shown by ANOVA and quadratic trend analysis. Data are presented as a) mean±sem and b–d) individual values with means shown as x.
ANOVA of CXCL10 concentrations in nasal lavage in seven out of 11 subjects showed significant differences over time (p<0.01), with a significant quadratic trend (p<0.01) (fig. 1b). Comparison of baseline and peak CXCL10 concentrations showed a significant mean (95% CI) increase of 247 (155–339) pg·mL−1 (p<0.01). ANOVA of CXCL10 concentrations in sputum showed significant differences over time (p<0.01), with a significant quadratic trend (p<0.01) (fig. 1c). Comparison of log-transformed peak against baseline concentrations showed a mean (95% CI) increase of 1.1 (0.5–1.6) log (p<0.01), i.e. a mean (95% CI) increase of 12-fold (3–40-fold). ANOVA of MxA, OAS, CXCL10 and IFN-β gene expression showed a significant difference over time (p<0.01, p<0.01, p=0.01 and p=0.04, respectively), with a significant quadratic trend for all assessments (p=0.01, p<0.01, p=0.04 and p=0.01, respectively). CXCL10 gene expression is shown in figure 1d as an example.
Ex vivo IFN-β gene expression in blood cells in response to poly(I:C) stimulation, assessed in 10 of the 11 subjects, was upregulated by a mean 75-fold, with a significant inverse correlation (Spearman’s correlation coefficient rs= -0.72, p=0.04) between the ex vivo induction of IFN-β and cold symptoms, but not with asthma symptoms or the biological response.
This is the first study to demonstrate the safety and feasibility of experimental RV infection in asthmatics requiring ICS, which paves the way for larger studies in more severe asthmatics. The increased asthma symptoms and antiviral responses in nasal lavage, sputum and blood suggest that such studies can be used to understand the disease mechanisms in this population for whom exacerbations present a greater risk than the type of asthmatic studied to date. Such studies also have the potential utility for testing new antiviral or anti-inflammatory drugs. However, we recognise that the mild nature of exacerbation induced is, to an extent, a limitation; thus, forced expiratory volume in 1 s did not drop significantly, possibly because of increased use of bronchodilators by an average of three puffs per day. Thus, this model cannot be used as a surrogate for more severe exacerbations and these will have to be studied in a natural setting on community-acquired exacerbations. Any experimental challenge in patients with asthma needs to demonstrate safety and the current study shows that this can be achieved while producing disease-relevant readouts.
To be valuable, experimental infection should induce significant changes in asthma-relevant end-points. The virus shedding and antiviral responses seen in sputum suggested spread of infection from the URT to the LRT. Virus shedding was temporally related to cold and increased asthma symptoms that were associated with increased bronchodilator use (from mean 0.4 to a peak of 3.2 times per day), which probably accounts for the lack of sustained fall in lung function. Unlike in steroid-naïve asthmatics [2], there was no significant change in bronchial hyperreactivity, possibly due to long-term ICS use [9]. Peak asthma symptoms followed peak cold symptoms by a mean 2.1 days. Correspondingly, URT and LRT symptoms were associated with nasal and sputum viral loads, peaking at post-inoculation days 3 and 5, respectively. Antiviral biomarkers in nasal lavage (CXCL10) and sputum (MxA, OAS and CXCL10) followed a similar pattern, presumably in response to infection. Interestingly, blood responses (MxA, OAS, CXCL10 and IFN-β genes) followed the pattern of nasal lavage and not sputum responses. Thus, a window of therapeutic opportunity is apparent between URT and LRT symptoms; accordingly, this model has utility for testing of therapies for virus-induced asthma exacerbations.
The increased susceptibility of the asthmatic lung to infection contributes to the association between virus infections and exacerbations [2, 10]. Therefore, we investigated whether ex vivo poly(I:C)-induced antiviral responses by whole blood cells related to clinical and biological outcomes. We found that antiviral gene expression correlated significantly with cold severity without predicting asthma symptom severity or the antiviral responses in the airways, possibly because the deficient lung epithelial responses to a virus play a more important role in defining how asthmatics respond to infection [10].
This study has limitations. First, there was no control group. Furthermore, we detected viral RNA by PCR without distinguishing between infectious and inactivated virus; however, detection of viral RNA and infectious virus has correlated closely in previous studies [2, 6]. Assessments of sputum and nasal lavage were possible in a small number of subjects; these observations should, therefore, be seen as preliminary. Finally, although all study participants were stable before infection, some of the observed changes could possibly be due to the variability that characterises asthma. Nevertheless, we believe that this study provides proof of concept for the use of an experimental virus infection model in the study of chronic asthmatics whose disease requires the use of ICS.
Acknowledgments
The authors wish to acknowledge all the staff, in particular the nurses, in the Southampton NIHR Biomedical Research Unit, the Southampton NIHR Wellcome Clinical Research Facility and the Imperial NIHR Comprehensive Biomedical Research Centre (all Southampton, UK) for their help with clinical aspects of the study, and the patient volunteers for donating their time and samples.
Footnotes
Support statement: This study was supported by a collaborative research grant from Synairgen plc (Southampton, UK).
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
- Received August 13, 2013.
- Accepted October 1, 2013.
- ©ERS 2014