



Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH
- Audrey Courboulin*,¶,
- Marjorie Barrier*,¶,
- Tanya Perreault*,
- Pierre Bonnet#⇓,
- Veronique L. Tremblay*,
- Roxane Paulin*,
- Ève Tremblay*,
- Caroline Lambert*,
- Maria H. Jacob*,
- Sandra N. Bonnet*⇓,
- Steeve Provencher* and
- Sébastien Bonnet*⇓
- *Dept of Medicine, School of Medicine, Laval University, Quebec; Institut universitaire de cardiologie et de pneumologie de Québec/IUCP Quebec, Canada
- #Faculty of Medicine, Dept of Medicine, Tours, France
- ¶These authors have contributed equally
- S. Bonnet, Centre de recherche, Institut universitaire de cardiologie et de pneumologie de Québec. E-mail: sebastien.bonnet{at}criucpq.ulaval.ca
Abstract
Like cancer, pulmonary arterial hypertension (PAH) is characterised by a pro-proliferative and anti-apoptotic phenotype. In PAH, pulmonary artery smooth muscle cell (PASMC) proliferation is enhanced and apoptosis suppressed. The sustainability of this phenotype requires the activation of pro-survival transcription factors, such as signal transducer and activator of transcription (STAT)3 and nuclear factor of activated T-cells (NFAT). There are no drugs currently available that are able to efficiently and safely inhibit this axis. We hypothesised that plumbagin (PLB), a natural organic compound known to block STAT3 in cancer cells, would reverse experimental pulmonary hypertension.
Using human PAH-PASMC, we demonstrated in vitro that PLB inhibits the activation of the STAT3/NFAT axis, increasing the voltage-gated K+ current bone morphogenetic protein receptor type II (BMPR2), and decreasing intracellular Ca2+ contentration ([Ca2+]i), rho-associated coiled-coil containing protein kinase (ROCK)1 and interleukin (IL)-6, contributing to the inhibition of PAH-PASMC proliferation and resistance to apoptosis (proliferating cell nuclear antigen (PCNA), TUNEL, Ki67 and anexine V). In vivo, PLB oral administration decreases distal pulmonary artery remodelling, mean pulmonary artery pressure and right ventricular hypertrophy without affecting systemic circulation in both monocrotaline- and sugen/chronic hypoxia-induced PAH in rats.
This study demonstrates that the STAT3/NFAT axis can be therapeutically targeted by PLB in human PAH-PASMC and experimental PAH rat models. Thus, PLB could be considered a specific and attractive future therapeutic strategy for PAH.
- Apoptosis
- plumbagin
- pulmonary arterial hypertension
- sugen
- vascular remodelling
Pulmonary arterial hypertension (PAH) is a devastating disease of the pulmonary vasculature defined by an increase in pulmonary artery pressure (Ppa) due to a sustained elevation of the pulmonary vascular resistance, which rapidly induces the failure of the right ventricle [1]. At the cellular level, PAH is characterised by enhanced inflammation [2], proliferation and resistance to apoptosis of the pulmonary artery smooth muscle cells (PASMCs) [3, 4]. The sustainability of this phenotype is due in part to the activation of the transcription factor signal transducer and activator of transcription (STAT)3 [5, 6]. This suggests that STAT3 inhibition could be of a great therapeutic interest for PAH. In a similar way to cancer, STAT3 activation in PAH has been associated with the upregulation of the oncogene provirus integration site for the moloney murine leukaemia virus (Pim-1), promoting the activation of the transcription factor of the nuclear factor of activated T-cells (NFATc2) [6]. NFATc2 activation has been shown to account for both proliferation and resistance to apoptosis in cancer and PAH [4, 7]. Indeed, by downregulating K+ channels, such as Kv1.5, NFATc2 causes cell depolarisation, increasing intracellular Ca2+ concentration ([Ca2+]i) and promoting cell proliferation; by upregulating Bcl-2, NFATc2 activation leads to mitochondrial hyperpolarisation and apoptosis resistance [4]. Moreover, STAT3 axis is implicated in tumoral upregulation of survivin [8, 9], described as an important protein in the pathogenesis of PAH [10]. Finally, STAT3 has also been associated with bone morphogenetic protein receptor type II (BMPR2) downregulation, further promoting the pro-proliferative, anti-apoptotic PAH phenotype [11]. For all these reasons, the STAT3/NFAT axis can be considered a major signalling hub for PAH.
Taken together, previous studies suggest that STAT3 axis inhibition might represent an attractive therapeutic strategy for PAH. Nevertheless, no clinically available drugs that target STAT3 axis are currently available.
Plumbagin (PLB), or 5-hydroxy-2-methyl-1,4-naphthoquinone, is a natural product found in the plants of the Plumbaginaceae, Droseraceae, Ancestrocladaceae and Dioncophyllaceae families. PLB has been shown to exert anticancer activities against a wide variety of tumour cells, including breast cancer and lung cancer, in which NFAT is activated [4, 12–15]. However, the mechanisms accounting for PLB anticancer effects remain unknown. Recent studies have shown that PLB downregulates the expression of survivin and growth factor receptor [16], which are implicated in PAH [9, 17] and modulated by STAT3 [18, 19] and NFAT [4]. More importantly, PLB was found to be an efficient STAT3 inhibitor in cancer cells [20], thus promoting apoptosis [21]. We therefore hypothesised that inhibition of the STAT3/NFAT axis by PLB would decrease PAH-PASMC pro-proliferative and anti-apoptotic phenotypes, thus preventing and reversing established PAH.
MATERIALS AND METHODS
All the experiments were performed with the approval of the Laval University Ethic and Biosafety Committee (Laval University, Quebec, Canada). The study was performed in accordance with the Guide for the Care and Use of Laboratory Animals [22]. It conforms to the principles outlined in the Declaration of Helsinki. All patients gave informed consent before the study (ethic committee protocol number 20142).
Cell culture
Human PAH-PASMCs were isolated from the ∼1,5 μm-diameter small pulmonary arteries of three PAH patients. Human healthy PASMCs (n=5) were purchased (#302K-05a; Cell Application, San Diego, CA, USA). PASMCs were grown in high-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS (Gibco, Invitrogen, Burlington, Canada) and 1% antibiotic/antimytotic (Gibco) [7] and used until the fifth passage. Plumbagin (PLB) was purchased from Sigma-Aldrich (Ontario, Canada) and dissolved in <1% of dimethyl sulfoxide (DMSO). For all of the experiments, PAH-PASMC and PASMC were treated for 48 h. Platelet-derived growth factor (PDGF; 30 ng·mL−1), endothelin (ET)-1 (10 nM) and angiotensin II (200 nM) were all from EMD Biosciences, Mississauga, Canada.
Immunofluorescence
Measurements of the mitochondrial membrane potential (ΔΨm) and [Ca2+]i in live PASMC (37°C) were performed using tetramethylrhodamine methyl-ester perchlorate (TMRM) and Fluo-3AM from Invitrogen (Branchburg, NJ, USA) at a final concentration of 5 μM, as previously described [4, 23]. TMRM and Fluo-3 fluorescence intensity were measured (20–50 cells per patient in three PAH and five healthy patients). PASMC apoptosis rates were measured using TUNEL and annexin V (Millipore, Temecula, CA, USA) after serum starvation (0.1% FBS in 48 h) and proliferation using Ki67 and the proliferating cell nuclear antigen (PCNA) antibody (DAKO, Carpinteria, CA, USA) according to the manufacturers' instructions [4, 23]. The percentage of cells that was nuclei positive for TUNEL, annexin V, PCNA or Ki67 was determined (20–50 cells per patient in three PAH and five healthy patients, or five arteries per animal in five animals). PY705-STAT3 (Cell Signalling, Danvers, MA, USA; diluted 1/250) and NFATc2 (Abcam, Cambridge, MA, USA; diluted 1/250) staining were performed as previously described [4]. The number of cells presenting a nuclear localisation of the protein was measured in 20–50 cells per patient in three PAH and five healthy patients, or five arteries per animal in five animals. Alexa Fluor 488 or 594 (Invitrogen; diluted 1/1,000) were used as secondary antibodies.
Quantitative RT-PCR
To measure NFATc2 and BMPR2 expression (Taqman Gene Expression Assay; Applied Biosystem, Foster, CA, USA), total mRNA was extracted from PAH-PASMC or control PASMC using trizol protocol, as previously described [4, 22]. 18S was used as housekeeping gene for quantitative RT-PCR.
Western blot
Total protein fraction was extracted from either PASMC or distal PA as indicated. PY705-STAT3 and STAT3 (both Cell signalling, diluted 1/1,000) were quantified and normalised to the smooth muscle actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; 1/400), as previously described [7]. BMPR2 (Abcam, diluted 1/1,000), rho-associated coiled-coil containing protein kinase (ROCK)1 (BD Biosciences, Mississauga, Canada; 1/1,000) and PKCϵ (Santa Cruz Biotechnology, diluted 1/100) were normalised to Ponceau S (Fisher Scientific UK, Loughborough, UK). Evaluation for PY705-STAT3/STAT3 were obtained from the same gel after striping (30 min at 50°C).
Electrophysiology
Standard whole-cell patch clamping was realised on voltage-clamped cells at a holding potential of -70 mV, with solutions permitting potassium current [23]. Currents were evoked using 200-ms test pulses from -70 mV to +70 mV, with 10-mV steps, as previously described (filtered at 1 kHz and sampled at 2–4 kHz) [23]. The results are shown with the current density, as the observed cell current was normalised with its own cellular capacity. The results are also shown with 4-AP sensitive current, representing the amount of K+ current Kv channels dependent, as 4-AP is a Kv channel blocker. PAH-PASMC or healthy PASMC were chronically treated with PLB (48 h; 1 μM) and with 4-AP (1 mM) during the patch clamping.
In vivo experiments
Male Sprague-Dawley rats were injected subcutaneously with 60 mg·kg−1 of monocrotaline (MCT; Sigma-Aldrich). PLB was given orally (gavage) at a concentration of 4 mg·kg−1, either every day for 4 weeks following MCT injection and before PAH establishment (prevention protocol; n=5), or every day for 2 weeks after PAH establishment (haemodynamic measurment), i.e 15 days after MCT injection (reversal protocol; n=5).
Male Sprague-Dawley rats were injected s.c. with 20 mg·kg−1 of sugen (#S8442-25MG; Sigma Aldrich) and maintained in hypoxia (10% oxygen (O2)) for 3 weeks. 2 weeks after the end of the hypoxia protocol, the rats were orally (gavage) treated with PLB at 4 mg·kg−1 for 2 weeks.
Haemodynamic measurements
All rats underwent haemodynamic and echocardiography measurements, as previously described [4]. Briefly, pulmonary arterial acceleration time (PAAT), known to decrease with the PAH severity in both rats and patients, was measured using echo Doppler [4, 24], and right ventricle thickness was measured via echocardiography. Right catheterisations (closed chest) were performed using science catheters.
Histology measurements
Pulmonary artery media wall thickness was assessed as previously described [4]. Briefly, paraffin lung sections were stained with haematoxylin–eosin, and pulmonary artery media wall thickness was measured using Image ProPlus software (Media Cybernetics, Bethesda, MD, USA). Two measurements per artery in five animals for each group were performed.
Data analysis
Averaged data are presented as the mean±sem. Normality of data was assessed by the Shapiro-Wilk normality test. All data were normally distributed. For comparison between two means, an unpaired t-test was used. ANOVA followed by the Dunn's post-test was used for comparison between more than two means. For correlation, a Pearson's test was performed.
RESULTS
PLB decreases PAH-PASMC proliferation and resistance to apoptosis
To study the effects of PLB on PASMC proliferation and apoptosis in vitro, human PAH-PASMCs were exposed to 10% FBS (a condition that is known to promote proliferation) or 0.1% FBS (a “starvation” condition that promotes apoptosis) [4, 7, 25]. PAH-PASMCs were treated with increasing doses (1 nM, 10 nM, 100 nM, 1 μM, 5 μM, 10 μM) of PLB or its proper vehicle (DMSO) (fig. 1a). Compared with healthy PASMCs, PAH-PASMCs have a greater proliferation rate and were more resistant to starvation-induced apoptosis. PLB dose-dependently decreases proliferation and promotes apoptosis. Based on the dose–response effects, we decided to use PLB at 1 μM (which also corresponds with the dose previously used in other studies [20]) and were able to significantly decrease both proliferation and resistance to apoptosis. At 1 μM, PLB in starved PAH-PASMCs (0.1% FBS) increased apoptosis measured by both TUNEL and annexin V when compared with vehicle-treated PAH-PASMCs (fig. 1). In PAH-PASMCs exposed to 10% FBS, PLB (1 μM) decreased around two-fold of the proliferation measured by PCNA and Ki67 (fig. 1). Moreover, PLB effects on apoptosis were confirmed by TUNEL in staurosporine-treated PASMCs (fig. s1a) [26, 27].
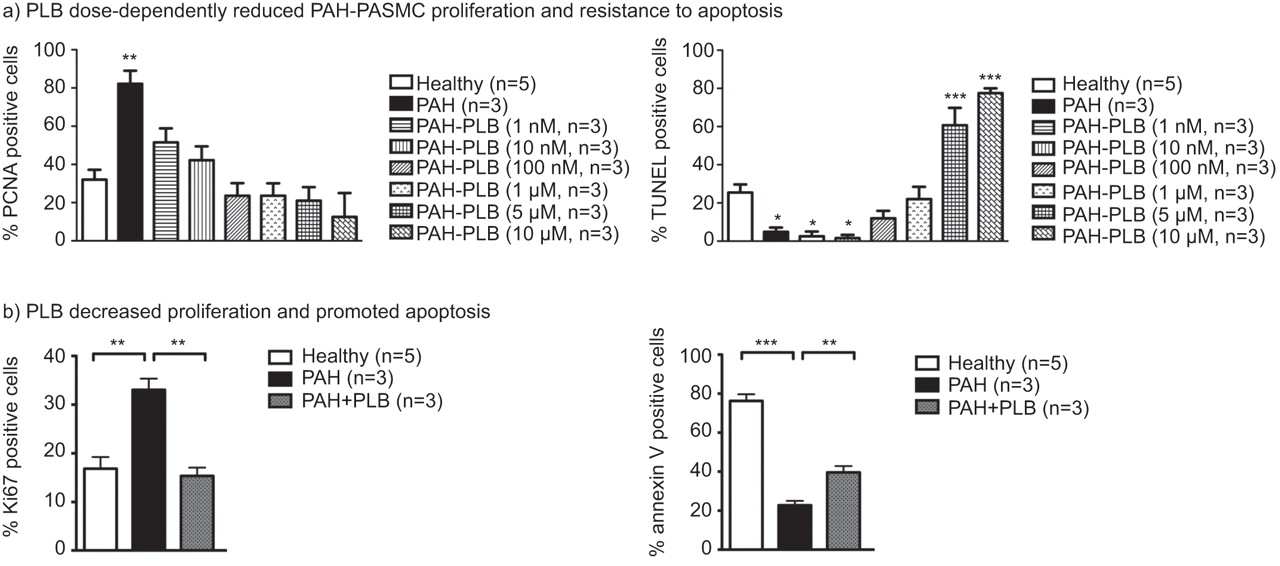
a) Proliferating cell nuclear antigen (PCNA) showed a significant increase in pulmonary arterial hypertension (PAH)-pulmonary artery smooth muscle cell (PASMC) proliferation compared with control-PASMC; plumbagin (PLB; 1 nM, 10 nM, 100 nM, 1 μM, 5 μM, 10 μM) dose-dependently decreased PAH-PASMC proliferation. TUNEL assay revealed a significant resistance to apoptosis in PAH-PASMCs compared with healthy PASMCs, which was dose-dependently decreased by PLB (1 nM, 10 nM, 100 nM, 1 μM, 5 μM, 10 μM). n=100–150 cells per patient. Given these results, the PLB concentration for the following experiments was chosen to be 1 μM. b) PLB (1 μM for 48 h) reversed PAH-PASMC proliferation (Ki67) and resistance to apoptosis (annexin V). n=100–150 cells per patient. *: p<0.05; **: p<0.01; ***: p<0.001.
The effects of PLB on proliferation were further confirmed in healthy PASMCs treated for 48 h with PDGF (30 ng·mL−1), angiotensin II (200 nM) and ET-1 (10 nM), all accepted STAT3 activators [28] and all known to be increased in PAH [29, 30] in presence and absence of PLB. As expected, PLB significantly decreases proliferation in PASMCs treated with Ang II, PDGF and ET-1 (Fig. s1b).
PLB decreases the STAT3/NFAT axis activation in PAH-PASMCs
We have previously shown that the pro-proliferative and anti-apoptotic phenotype seen in PAH-PASMCs was most often attributed to the activation of the STAT3/NFAT axis [4–6]. As PLB significantly decreases PAH-PASMC proliferation and resistance to apoptosis, we measured whether PLB affects STAT3 and NFATc2 activation in PAH-PASMCs. STAT3 and NFATc2 activations were measured in human PASMCs isolated from control and non-familial PAH patients by quantitative RT-PCR (NFATc2), immunoblot (PY705-STAT3/STAT3) and nuclear translocation assay (confocal microscopy). Compared with control PASMCs, the PY705-STAT3/STAT3 ratio was increased around two-fold in PAH-PASMCs (fig. 2a), which was associated with a greater nuclear translocation (fig. 2b). Similarly, an approximately two-fold increase in NFATc2 expression and nuclear translocation was also observed in PAH-PASMCs compared with controls (fig. 2). PLB (1 μM for 48 h) decreased both STAT3 and NFAT activation in PAH-PASMCs. These findings suggest a significant activation of STAT3 and NFATc2 in PAH-PASMCs compared with control-PASMCs, which can be inhibited by PLB (fig. 2b).

Plumbagin (PLB) reverses STAT3/nuclear factor of activated T-cells (NFAT) in human pulmonary arterial hypertension (PAH)-pulmonary artery smooth muscle cells (PASMCs). a) NFATc2 expression was quantified by RT-PCR in human healthy and PAH-PASMCs. mRNA expression was normalised to 18S. As shown, NFATc2 expression significantly increased in PAH-PASMCs compared with control-PASMCs. PLB (1 μM for 48 h) significantly decreased NFATc2 expression. PY705-STAT3/STAT3 ratio, accounting for STAT3 activation, was assessed by immunoblot. As shown, STAT3 was upregulated in PAH-PASMCs compared with control-PASMCs, and PLB (1 μM for 48 h) reversed this activation. n=3 experiments per patient. b) NFATc2 and STAT3 activation were further assessed in PASMCs using nuclear translocation assay. As shown, the percentage of NFATc2 and PY705-STAT3 translocated to the nucleus significantly increased in PAH-PASMCs compared with healthy. PLB (1 μM for 48 h) significantly decreased both NFATc2 and STAT3 activation. n=100–150 cells per patient. DAPI: 4′,6-diamidino-2-phenylindole. *: p<0.05; **: p<0.01.
PLB reverses the activation of pathophysiological pathways affected by the activation of the STAT3/NFAT axis
In PAH-PASMCs, STAT3/NFAT-mediated proliferation [4, 31] has been linked with the downregulation of voltage-gated K+ (Kv) channels [32, 33], resulting in membrane depolarisation [31, 33], the opening of voltage-dependent calcium channels, and an increase in [Ca2+]i [4, 31, 34]. Using whole-cell patch clamping, we demonstrated that PLB (48 h) restores the decrease in total K+ current density observed in PAH-PASMCs (fig. 3a and b ). This increase in K+ current was attributed to the activation of the Kv channels, as this increase was totally blocked by 4-AP, a specific Kv channel blocker (fig. 3a and c) [35]. It is noteworthy that PLB has no effect on healthy PASMCs. These results are consistent with previously published studies [4, 36], demonstrating that the STAT3/NFAT axis is implicated in Kv channel regulation such as Kv1.5.

Plumbagin (PLB) reverses K+ current density decrease in pulmonary arterial hypertension (PAH)-pulmonary artery smooth muscle cells (PASMCs) through the increase of 4-AP-sensitive current density. a) Representative K+ currents with or without PLB (1 μM for 48 h) and 4-AP (1 mM) in healthy and PAH-PASMCs. b) PAH-PASMCs show a significant decrease in K+ current density compared with vehicle-treated PASMCs. PLB (1 μM for 48 h) increases PAH-PASMC current density to a level similar to the one seen in healthy PASMCs. c) The increased current density mainly relies on an upregulation of the voltage-gated K+ channel (Kv) current as shown by a greater sensitivity to the Kv channel blocker 4-AP at 70 mV. ns: nonsignificant. *: p<0.05; **: p<0.01.
Using the Fluo-3AM technique, we investigated whether the increases in Kv current caused by PLB decrease [Ca2+]i. As expected, PLB decreases [Ca2+]i in PAH-PASMCs to a level similar to that seen in healthy PASMCs (fig. 4a).

Plumbagin (PLB) decreased intracellular Ca2+ concentration ([Ca2+]i) in pulmonary arterial hypertension (PAH)-pulmonary artery smooth muscle cells (PASMCs) and mitochondrial hyperpolarisation; it reversed bone morphogenetic protein receptor type II (BMPR2) and rho-associated coiled-coil containing protein kinase (ROCK)1 downregulation, and interleukin (IL)-6 upregulation. a) PAH-PASMCs showed a significant increase in [Ca2+]i compared with healthy PASMC, which was reversed PLB (1 μM for 48 h). PAH-PASMCs displayed mitochondrial membrane potential (ΔΨm) hyperpolarisation compared with healthy PASMCs. PLB (1 μM for 48 h) caused a significant ΔΨm depolarisation (decreased tetramethylrhodamine methyl-ester perchlorate (TMRM), red fluorescence) compared with vehicle-treated PAH-PASMCs. b) Compared with healthy PASMCs (n=5 patients), PAH-PASMCs (n=3 patients) showed a significant decrease in BMPR2 expression (quantitative RT-PCR and Western blot, n=3 experiments) and an upregulation in ROCK1 (Western blot, n=3), both of which were reversed by PLB (1 μM for 48 h). Finally, PLB (1 μM for 48 h) significantly decreased IL-6 expression (quantitative RT-PCR, n=3) in PAH-PASMCs. FU: fluorescence units. *: p<0.05; **: p<0.01; ***: p<0.001.
Because STAT3 and NFAT are implicated in ΔΨm regulation [7, 37], the increase in apoptosis following PLB treatment might result from the activation of mitochondrial-dependent apoptosis. As the mitochondria transition pore is voltage-dependent [38], the ΔΨm depolarisation is a threshold index for mitochondrial-dependent apoptosis. In fact, apoptosis is associated with decreased ΔΨm. To investigate the mechanism by which PLB promotes apoptosis, we measured ΔΨm using TMRM in PAH-PASMCs treated with PLB. PLB caused a significant ΔΨm depolarisation (decreased TMRM, red fluorescence) compared with vehicle-treated PAH-PASMCs. These data confirmed that PLB affects the ΔΨm and therefore enhances mitochondrial-dependent apoptosis (fig. 4a).
In addition to both the Kv/calcium axis and ΔΨm, other pathways are known to be implicated in PAH-PASMC proliferation and resistance to apoptosis, including: BMPR2 protein and mRNA expression [39]; ROCK1 activation [40]; and mRNA interleukin (IL)-6 expression [41]. Although the importance of each pathway in the aetiology of PAH remains to be established (and is likely to be different among the different forms of PAH), we provide evidence that PLB can affect all of them. Indeed, PLB significantly increased BMPR2 expression and decreased IL-6 and ROCK1 in PAH-PASMCs (fig. 4b). Although the exact molecular mechanism by which PLB affects these pathways remains unknown, the fact that all of them have been shown to be affected by STAT3 reinforces the concept that the STAT3/NFAT axis is an important integrative signal hub in PAH and that its inhibition might represent a novel and effective therapeutic strategy to improve PAH.
PLB reverses experimental PAH in rats
To determine the putative therapeutic potential of PLB, we determined in vivo whether PLB (4 mg·kg−1·day−1 per os) can reverse establish PAH, in two accepted experimental rat PAH models: the monocrotaline (MCT) induced-PAH and the sugen/chronic hypoxia models. Longitudinal studies to assess the efficiency of our treatments were performed using noninvasive measurements (exercise capacity test; Doppler and echocardiography) [4]. In the prevention MCT model, PLB prevented changes in pulmonary haemodynamics, right ventricle free wall thickness, and general cardiac function (exercise capacity on treadmill) seen in the vehicle-treated rats. When given in rats with established PAH (2 weeks post-MCT injection to reverse the disease), PLB was shown to increase PAAT (a Doppler parameter known to correlate well with Ppa in both humans and rats; as the Ppa rises, PAAT shortens) [4, 42], decrease right ventricle wall thickness and increase exercise capacity (fig. 5a). These findings were invasively confirmed by direct PA pressure measurements in close chest animals (fig. 5b). Interestingly the cardiac output was not affected by PLB treatment (fig. 5b).

Plumbagin (PLB) prevented and reversed pulmonary arterial hypertension (PAH) in rats injected with monocrotaline (MCT). The effects of orally delivered PLB (4 mg·kg−1·day−1) in the prevention protocol (PLB treatment beginning the same day as MCT injection, before PAH establishment) for 4 weeks or in the reversion protocol (PLB treatment beginning 2 weeks post MCT injection once PAH was established) for 2 weeks were longitudinally assessed for 4 weeks. a) As shown, PLB prevented and reversed PAH (n=5 rats per group), assessed noninvasively by Doppler echocardiography (pulmonary arterial acceleration time (PAAT)), M-mode ultrasound (right ventricular (RV) hypertrophy), and exercise capacity (treadmill). n=5 rats for each group. b) These findings were invasively confirmed by direct pulmonary artery pressure (Ppa) measurements. n=5 rats for each group. Note that PLB did not affect cardiac output. c) PLB treatment significantly prevented and reversed distal pulmonary artery (<300 μm) remodelling measured by haematoxylin–eosin (H&E) staining. n=10 arteries per rat with five rats per condition. *: p<0.05; **: p<0.01; ***: p<0.001.
In order to determine whether PLB would reduce pulmonary artery remodelling in MCT-PAH rats, we measured pulmonary artery media wall thickness using haematoxylin–eosin staining on lung histological sections. PLB given in prevention or in attempt to reverse established PAH in rats showed a significant reduction in the percentage medial thickness in small and medium-sized pulmonary arteries (fig. 5c). This finding was associated with a significant decrease in STAT3 and NFATc2 activation (fig. 6a), and also in Sarcoma (Src) activation (fig. 6b), which we have previously shown to be the pathway activating STAT3 in PAH-PASMC [5], decreasing PASMC proliferation (Ki67) and resistance to apoptosis (TUNEL) in vivo (fig. 6c and fig. s2a).

Plumbagin (PLB) decreased STAT3, nuclear factor of activated T-cells (NFAT)c2 and Sarcoma (Src) activation, decreasing the associated pulmonary artery smooth muscle cell (PASMC) proliferation and resistance to apoptosis seen in the distal pulmonary arteries. a and b) PLB-treated animals had a significant reduction in STAT3, NFATc2 and Src activation in the distal pulmonary arteries measured by immunofluorescence (IF), compared with the distal pulmonary arteries of untreated rats. n=5 arteries per rat with five rats in each condition. c) Decrease in PASMC proliferation (Ki67; n=5 arteries per rat in five rats; p<0.05) and resistance to apoptosis (TUNEL n=5 arteries per rat in five rats; p<0.05) both in prevention and reversion protocol. PAH: pulmonary arterial hypertension; DAPI: 4′,6-diamidino-2-phenylindole; P-Src: phosphorylated Src; FU: fluorescence units; MCT: monocrotaline. *: p<0.05; **: p<0.01; ***: p<0.001.
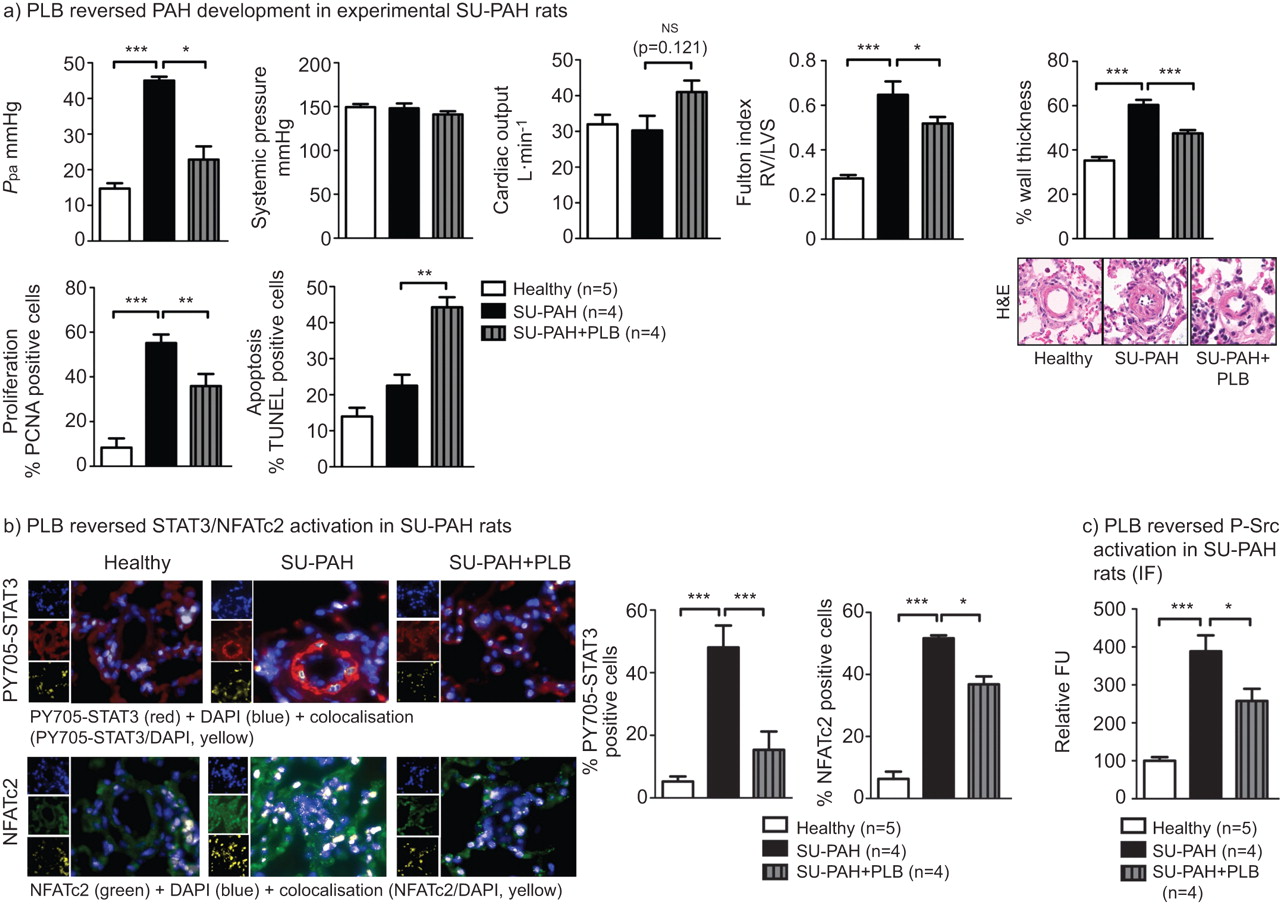
Although the MCT-induced PAH model is a very robust model of PAH, to further demonstrate the therapeutic effect of PLB, we tested whether PLB improved PAH in rats injected (s.c.) with sugen, a type 2 vascular endothelial growth factor receptor antagonist [43, 44], exposed to chronic hypoxia (10% O2) for 3 weeks, and placed back under normoxic conditions for an additional 4 weeks. PLB (4 mg·Kg−1·day1 per os) was given once PAH was established (5 weeks post sugen injection) for 2 weeks.
PLB significantly decreased mean Ppa, right ventricular hypertrophy and distal pulmonary artery remodelling, though in a less efficient way than in the MCT model (fig. 7a). As expected, these effects were associated with decreased distal pulmonary artery PASMC proliferation and increased apoptosis (fig. 7a and Fig. s2b), associated with a significant decrease in STAT3 and NFATc2 activation measured by nuclear translocation assay in the distal pulmonary arteries (<300 μm; fig. 7b), and with a significant decrease in Src activation (fig. 7c).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo, in sugen (SU)-induced rats, plumbagin (PLB) administration reverse established pulmonary arterial hypertension (PAH). The effect of orally delivered PLB (4 mg·kg−1·day−1) was assessed in the SU/chronic hypoxia rat model. a) PLB reverses PAH, assessed by direct pulmonary artery pressure (Ppa) measurements, without significantly affecting systemic pressure or cardiac output (n=5 rats per group). Furthermore, PLB reverses right ventricular (RV) hypertrophy, assessed invasively by the Fulton index (n=5 rats for each group). The remodelling has also been assessed by haematoxylin–eosin (H&E) staining (n=10 arteries per rat with five rats per condition). PLB treatment significantly reverses distal pulmonary artery (<300 μm) remodelling. These results are associated with a PLB-dependant decrease in proliferation and an increase in apoptosis in SU-treated rats. b and c) PLB treatment is associated with a significant decrease in both STAT3, nuclear factor of activated T-cells (NFAT)c2 and Sarcoma (Src) activation (n=5 arteries per rat with five rats in each condition). ns: nonsignificant; LVS: left ventricular+septum; PCNA: proliferating cell nuclear antigen; DAPI: 4′,6-diamidino-2-phenylindole; P-Src: phosphorylated Src; IF: immunofluorescence; FU: fluorescence units. *: p<0.05; **: p<0.01; ***: p<0.001.
DISCUSSION
We have provided, for the first time, evidence showing that PLB decreases the STAT3 constitutive activation seen in PAH. We have shown that PLB-dependant STAT3 inhibition reverses PAH phenotype both in vitro and in vivo. Indeed, PLB restores most of the molecular and cellular abnormalities seen in human PAH-PASMC, including decreased activation of NFATc2, STAT3, IL-6 and ROCK1, and upregulation of BMPR2 (figs 2 and 4). All of these demonstrated pathways contribute to the decrease in human PAH-PASMC proliferation and resistance to apoptosis. Using two independent experimental models of PAH (MCT and sugen/chronic hypoxia) in vivo, we provide strong evidence that PLB is able to reverse PAH, highlighting its possible efficiency to treat this lethal pathology in PAH patients. Indeed, in both models, PLB significantly improves Ppa, pulmonary artery medial hypertrophy and right ventricular hypertrophy, without affecting systemic pressure and cardiac output (figs s5, s6 and s7).
Although previous studies have reported a putative inotropic effect of PLB in the isolated heart model, our in vivo study with PLB given orally did not significantly affect global cardiac function, such as cardiac output [45, 46]. Moreover, it was shown to significantly improve exercise tolerance in rats with PAH, without affecting systemic pressure (figs 5b and 7a). Although several other measurements are needed to totally exclude long-term toxicity or adverse effects, our findings open up a new avenue of investigation and support a putative therapeutic role for PLB in PAH.
We are the first group to report an improvement of PAH in the sugen/chronic hypoxic rat model. The efficiency of PLB in two independent PAH models, such as the MCT and the sugen models, is a true indication that PLB is of great therapeutic interest. Moreover, we demonstrated in both models that, as in human PAH-PASMCs, both STAT3 and NFAT are upregulated in the distal PA and their inhibitions significantly improve PAH in the sugen-treated rats; this strongly reinforces the importance of this pathway in the aetiology of PAH.
The activation of the STAT3/NFAT axis that we described is likely to have a multifactorial aetiology in PAH. Indeed, both STAT3 and NFAT may be critical integrators of multiple signalling pathways, and their downstream effects could explain several important features of PAH. This could explain why STAT3 inhibition may be efficient in reversing PAH. In vivo, endothelial dysfunction and inflammation are recognised as some of the earliest abnormalities in PAH, resulting in a well-recognised imbalance of endothelium-derived vasoactive factors, with increased vasoconstrictors (endothelin [47], thromboxane [48]), all of which lead to STAT3 activation and decreased vasodilators (such as nitric oxide or prostacyclin [48]). In addition, increased circulating growth factor [49] and cytokines [50] have been reported in PAH, which also activate STAT3. Thus, it is likely that circulating factors are implicated in STAT3 activation in PAH. Due to the presence of the STAT3 binding sequence within the NFATc2 promoter region [51], the increase in NFATc2 expression seen in PAH-PASMCs has been attributed to STAT3 activation [5, 6]. Moreover, STAT3 positively regulates the NFAT activator Pim1, which could explain the increase in NFAT activation [10]. Once activated, NFAT regulates multiple genes, which might positively reinforce its own expression and activation. For example, the downregulation of Kv1.5 leads to PASMC depolarisation, opening of L-type Ca2+ channels and a sustained increase in [Ca2+]i, (as shown in our study), and thus calcineurin-dependent NFAT activation [4]. NFAT is not the only mechanism affected by STAT3 in the PAH phenotype. Indeed, it has been recently proposed that STAT3 activation upregulates microRNAs (miR-17/92), accounting for the downregulation of the receptor BMPR2 in PASMCs [11]. Moreover, several STAT3-related proteins have been implicated in RhoA/ROCK activation [52], along with IL-6 expression [11]. This not only demonstrates the importance of the STAT3 axis in PAH but also shows that STAT3 can be considered as an integrator of the multiple pathways implicated in PAH, including NFAT activation, BMPR2 downregulation RhoA/ROCK activation and IL-6 expression [39, 53]. We therefore believe that the PLB-dependant inhibition of STAT3 could explain how a single drug such as PLB can affect so many pathophysiological pathways and be efficient in several experimental models (figs 5, 6 and 7).
We are not the first group to report the beneficial health effects of PLB. In fact, this natural organic compound has been previously shown to have anti-proliferative and pro-apoptotic properties (in part, through STAT3 inhibition [20]) in cancer and vasoactive properties in bovine pulmonary arteries [54], all of which agree with a positive therapeutic effect in PAH. In this study, we have demonstrated an inhibitory effect of PLB on STAT3 activation in human PASMCs and shown that it also blocks NFAT expression and activation. Both effects induce ΔΨm depolarisation and decrease [Ca2+]i through the upregulation of Kv channels, as previously described [4, 37]. These effects are likely to be responsible for the inhibition of PAH-PASMC proliferation and resistance to apoptosis in vitro and in vivo in the reduction of the distal pulmonary artery remodelling processes. Indeed, we and others have previously reported that increasing Kv channels, such as Kv1.5 in PAH-PASMC, is sufficient to decrease [Ca2+]i and PASMC proliferation [34, 55], while depolarising mitochondria [9, 56] is sufficient to promote apoptosis in PAH-PASMCs. As both mechanisms are controlled by STAT3 and NFAT [4, 6], the inhibition of the STAT3/NFAT axis by PLB should promote PAH-PASMC apoptosis and decrease proliferation. This is supported by our findings.
The mechanism of action of PLB remains elusive. Whereas it is likely that the mechanisms leading to NFAT inhibition by PLB rely on STAT3 inhibition [5, 6], the mechanism accounting for STAT3 inhibition remain to be established. We recently showed that JAK2 (one of the most important STAT3 regulators [57]) is not implicated in PAH-PASMCs [5]; thus, the effect of PLB is unlikely to be mediated by JAK2 inhibition. The second regulator of STAT3 activation is the Src pathway [58]. Src has been shown to be implicated in PAH [5, 31] and in the activation of STAT3 in human PAH-PASMCs [5]. Although the implication of Src in STAT3 activation in human PAH-PASMCs has not been reassessed in the present study, we showed that Src is activated in the distal pulmonary arteries of both MCT and sugen rats, confirming our previous findings in humans. Moreover, we have demonstratede that Src activation is decreased in PLB-treated animals (figs 6b and 7c). Thus, inhibition of the Src pathway by PLB, as shown in cancer cells [20], could explain the STAT3 inhibition by PLB seen in PAH.
As STAT3 is an integrator of multiple pathways implicated in PAH, such as NFAT, BMPR2 and ROCK, it is not surprising that its inhibition by PLB is sufficient to improve PAH in both MCT and sugen models. Although several studies have shown that PLB can also affect many other pathways, including Akt or protein kinase (PK)Cε, which are implicated in systemic vascular remodelling processes [22, 59], their implication in PAH nonetheless remains elusive. For example, Akt is not activated in PAH [6]; thus, its putative inhibition by PLB should not affect PAH. The role of PKCε in PAH has been suggested, particularly in the contractile response and endothelial cell physiology; for example, PKCε inhibition decreases acute hypoxic vasoconstriction [60]. Nevertheless, PKCε knockout mice have a greater increase in right ventricular ystolic pressure, and right ventricular mass in response to chronic hypoxia than PKCε(+/+) mice [61]. Thus, if PLB effects were mediated by PKCε inhibition, PLB should worsen PAH and not improve it as shown in our study (fig. s3). Although the implication of other pathways cannot be ruled out, we believe that since PLB can affect most of the pathophysiological pathways implicated in PAH (STAT3, NFAT, BMPR2, ROCK, IL-6), it is already clinically very appealing.
Several other studies are needed to test the exact mechanism of action of PLB along with putative toxicity effects (no side-effects were observed in treated animals). Nevertheless, our findings provide strong evidence that such investigations are needed to eventually propose PLB as a new therapeutic tool for PAH. We believe that it will also lead to a better understanding of the regulation of apoptosis and proliferation by PLB, which will benefit many other human diseases, such as cancers.
Acknowledgments
The authors wish to thank C. Couture, C. Racine and S. Biardel (Laval University/Laval Hospital tissue bank, Institut universitaire de cardiologie et de pneumologie de Quebec, Quebec, Canada).
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
S. Bonnet has been funded for this work by the Canadian Institutes of Health Research (CIHR). S. Bonnet holds the position of Canada Research Chair. M.H. Jacob was supported by CAPES (the Brazilian Research Agency). A. Courboulin and R. Paulin are recipients of graduate scholarships from La Société Qébécoise d'Hypertension Artérielle (SQHA).
Statement of Interest
None declared.
- Received May 18, 2011.
- Accepted December 15, 2011.
- ©ERS 2012