



Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor
- *Division of Pharmacology and Pathophysiology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Sciences, Utrecht University, Utrecht, The Netherlands
- #Dept of Clinical Biochemistry, Faculty of Medical Sciences, Tarbiat Modares University, Tehran
- ¶Dept of Basic Science, Section of Biochemistry, Faculty of Veterinary Medicine, Urmia University, Urmia, Iran
- +Airways Disease Section, National Heart and Lung Institute, Imperial College London, London, UK
- E. Mortaz, Division of Pharmacology and Pathophysiology Utrecht Institute for Pharmaceutical Sciences Faculty of Sciences, Utrecht University, P.O. BOX 80082, 3508 TB Utrecht, The Netherlands. E-mail: emortaz{at}gmail.com
Abstract
Chronic obstructive pulmonary disease (COPD) is a major health problem and cigarette smoke is the main risk factor for the development of COPD. The characteristic changes in airway morphology, inflammatory cell infiltration and mediator expression in COPD may result from direct effects of cigarette smoke on airway cells. Toll-like receptors (TLRs) are key elements in pathogen recognition by the host immune system. Although TLRs have been intensely studied in innate immunity and infection, their critical role in noninfectious challenges has only recently emerged.
Here we investigate whether cigarette smoke induces TLR9 signalling in human neutrophils. Human neutrophils were isolated from buffy coat and exposed to cigarette smoke extract. The production of CXC chemokine ligand (CXCL)8 was measured as a functional readout and the role of TLR9 signalling was investigated. Cigarette smoke extract induced CXCL8 release via TLR9 activation in neutrophils, which was confirmed in TLR9 stably transfected human embryonic kidney 293 cells. Moreover, cigarette smoke extract upregulated the expression of TLR9 and the upregulated expression was suppressed by N-acetylcysteine.
TLR9 mediates cigarette smoke-induced release of CXCL8 and this may contribute to the accumulation of neutrophils and inflammation within the airways of smokers.
- Cigarette smoke
- neutrophils
- Toll-like receptors
Chronic obstructive pulmonary disease (COPD) is recognised as a major cause of death worldwide and poses an increasing global healthcare problem 1. As previously stated 2, the definition of COPD recognises the “abnormal”, exaggerated or amplified inflammatory response in the lung and systemically to cigarette smoking. The pattern of inflammation involves recruitment of lymphocytes, macrophages and neutrophils, as well as activation and damage to structural cells following the release of inflammatory chemokines and cytokines 2–5. In the Western world, the major driver of disease is cigarette smoke which is a complex mixture of organic chemicals, heavy metals and reactive oxygen species (ROS) 6–11. Importantly, Sopori 12 highlighted that chronic inhalation of cigarette smoke can modulate both innate and adaptive immune responses. Moreover, it has been speculated that many of the health consequences of chronic inhalation of cigarette smoke might be due to its adverse effects on the immune system 13.
It is likely that smokers develop airway inflammation through oxidative stress. Cigarette smoke activates macrophages and neutrophils to release proinflammatory mediators, chemokines and elastolytic enzymes 14. CXC chemokine ligand (CXCL)8 is an important chemokine produced by macrophages, neutrophils and epithelial cells and induces the recruitment of neutrophils to the airways 15, 16. As a part of the innate immune response, pattern recognition receptors mediate the interaction between conserved patterns on micro-organisms and the host. Toll like receptors (TLRs) are pathogen-associated molecular-pattern receptors for diverse microbially derived molecules expressed predominantly on innate immune cells 17. To date, 11 TLR family members have been identified in the human genome 18, 19. Bacterial DNA containing unmethylated CpG motifs act as important regulators of human neutrophil functions via TLR9. For example, stimulation of the TLR9 pathway via CpG oligonucleotides (ODN), induces CXCL8 production by neutrophils via the generation of peroxynitrite (ONOO-) 20.
Recently, we and others have demonstrated that cigarette smoke extract (CSE) activates TLR4 signalling 17, 21, 22. Furthermore, TLR4 seems to play a critical role in the development of lung emphysema 23. In the current study, we investigated whether CSE could also regulate TLR9 signalling in neutrophils. Thus, the present study was conducted to clarify TLR9 activation and expression in human primary neutrophils in response to CSE using the production of CXCL8 as a functional readout for TLR9 activation. Moreover, the involvement of ROS and a major signal transduction pathway (nuclear factor (NF)-κB) were investigated by the use of pharmacological inhibitors. The activation of NF-κB by CSE via TLR9 activation was also determined using genetic methods by overexpression of TLR9 in receptor-deficient cells and the subsequent analysis of NF-κB reporter gene activity.
MATERIALS AND METHODS
Reagents
Lipopolysaccharides (Escherichia coli 055.B5), N-acetylcysteine (NAC), curcumin, chloroquine, propidium iodide and NG-nitro-l-arginine methyl ester (l-NAME) were purchased from Sigma (Sigma-Aldrich, Zwijndrecht, The Netherlands). Roswell Park Memorial Institute (RPMI) 1640, Tyrode's buffer, fetal calf serum (FCS), nonessential amino acids, 2',7'-dichlorfluorescein-diacetate (DCFH-DA) (D399) and diaminofluorescein diacetate (DAF) were purchased from GibCo BRL Life Technologies (GIBCO-BRL-Invitrogen Corporation, Carlsbad, CA, USA). Rabbit polyclonal antibody against TLR9 was obtained from Santa Cruz Biotechnology (Tebu-bio, Heerhugowaard, The Netherlands). The precision protein standards and polyvinylidene fluoride (PVDF) membrane were purchased from Bio-Rad (Bio-Rad Laboratories, Veenendaal, The Netherlands). Horseradish peroxidase (HRP)-conjugated rabbit-anti mouse immunoglobulin (Ig)G and goat anti-rabbit IgG were purchased from Dako Diagnostics (Dako B.V. Heverlee, Belgium). CpG ODN 2395 stimulatory oligonuleotide, negative blocking control inhibitory ODN without the CpG motif (inhibitory ODN with sequence 5′-tttagggttagggttagggttaggg-3, ODN control (ODN 2395 control) with sequence 5′-tgctgcttttggggggcccccc-3′, Blasticidin S and QUANTI-Blue™ reagent were purchased from Invivogen (Cayla-InvivoGen Europe, Toulouse, France). Anti-tumour necrosis factor receptor-associated factor 6 (anti-TRAF6) (catalogue number sc-7221) and anti-interleukin (IL) receptor-associated kinase-1 Ab (catalogue number 06-872) antibodies were purchased from Santa Cruz Biotechnology and Upstate (Haarlerbergweg, The Netherlands), respectively. Bicinchoninic acid (BCA) protein assay kit and super-blocking buffer were purchased from Pierce (Perbio Science B.V, Etten-Leur, The Netherlands).
Protein A-Sepharose bead slurry and ECL or ECL Plus were purchased from Amersham Biosciences (Buckinghamshire, UK). TRIzol reagent and p-nifty 2×NF-κB secreted embryonic alkaline phosphatase (SEAP) plasmid and Sybergreen Universal PCR Master Mix and SuperScript III reverse transcriptase were purchased from Invitrogen (Carlsbad, CA, USA). Trans-AM NF-κB p65/NF-κB p50 Transcription Factor Assay Kit (Active Motif, Rixensart, Belgium). Phycoerythrin (PE)-labelled TLR9 and isotype IgG control were purchased from ebioscience (San Diego, CA, USA).
Preparation of CSE
Cigarette smoke-conditioned medium was produced as previously described 17. CSE was generated by the burning of commercially available cigarettes without filter, with tar 12 mg, nicotine 0.9 mg and CO 9 mg, using the TE-10z smoking machine (Teague Enterprises, Davis, CA, USA), which is programmed to smoke cigarettes according to the Federal Trade Commission protocol (35-mL puff volume drawn for 2 s, once per minute). Briefly, this machine was used to direct main- and side-stream smoke from one cigarette through a 5-mL culture medium (RPMI without phenol red). Hereafter, absorbance was measured spectrophotometrically at 320 nm and this number was taken as 100%. The pH of the resultant extract was titrated to pH 7.4 with medium, to different concentrations (0.75–3%). A 1.5% solution was used in the present study following preliminary experiments that indicated that this was non-toxic (viability ≥96%) and demonstrated a good induction of CXCL8 release 24.
Isolation of neutrophils
Human neutrophils were isolated from healthy volunteers as previously described 25. Briefly, human neutrophils were obtained from heparinised venous blood buffy coat by Ficoll–Hypaque centrifugation, followed by sedimentation in 5% dextran/0.9% saline. Neutrophils were separated from erythrocytes by lysis in a solution of 0.15 M NH4Cl, 0.01 M NaHCO and 0.01 M tetra-EDTA. The recovered neutrophils were resuspended in RPMI 1640 medium supplied with 10% fetal calf serum and essential amino acids and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.2, and washed three times. The purity of neutrophils preparations was >93–95%, as determined by Wright's staining of cytospin preparations. Cell viability of these cells was 97%, as determined by trypan blue exclusion. Neutrophils were kept on ice until used. For each experiment, n represents the number of separate individual donors used for each individual study.
Cell lines
TLR9 stably transfected human embryonic kidney (HEK) 293 cell lines (293XL-hTLR9, catalogue number 293xl-htlr9 for TLR9) and null HEK 293 (293-null, catalogue number 293-null) were purchased from Invivogen. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplied with 10% FCS and 10 mg·mL−1 of the antibiotic, blasticidin S, each for 3 days.
Cell activation
Neutrophils (5×106 cells·mL−1) were incubated for 20–30 min with l-NAME (0.1–10 mM) or NAC (0.1–10 mM); chloroquine (1–50 mg·mL−1) (enhances endosomal pH and thereby blocks lysomal pathways and used here as a blocker of TLRs signalling) 26 or blocking anti-sense ODN (1–10 mM) and then stimulated with CSE (1.5%), CpG ODN type A (3 μM) or control ODN (5 mM) for 30 min for western blot analysis. For CXCL8 determination 3×106cells·mL−1 were incubated for 9 h according to the conditions for Western blot analysis. For determination of intracellular CXCL8 expression, HEK 293 stably transfected TLR9 or null cells were stimulated with CSE or CpGODN for 6 h, and the whole cells extracts were subjected for western blot analysis. For preparation of samples for RT-qPCR, cells were activated for different time points with CSE or CpG ODN as described. The viability of cells before and after each experiment was determined by staining with propidium ionide or 7-ADD labelled with fluorescein isothiocyanate using flow cytometery (fluorescence-activated cell sorting (FACS) analysis).
Quantification of CXCL8 and cytokine assay
CXCL8 concentrations in cell supernatants were quantified using ELISA (BD Biosciences Pharmingen, Breda, The Netherlands) according to the manufacturer's instructions. The production of other inflammatory cytokines (tumour necrosis factor (TNF)-α, IL-6) was also measured in culture medium using CBA Kits (BD Biosciences Pharmingen) and flow cytometry (FACScalibur; BD Biosciences Pharmingen) according to the manufacturer's instructions.
Measurement of intracellular ROS stress and NO
Cells (1×106 cells·mL−1) were activated with CSE (1.5%) or CpG ODN (3 μM) for 1 and 5 h. After two washes with cold PBS, cells were pre-incubated with 10 μM redox-sensitive dye DCFH-DA (D399) for measurement of ROS and 5 μM of DAF for determination of NO for 20 min as previously described 27, 28. Intracellular levels of ROS and NO were determined by flow cytometry (FACScalibur). The data were plotted and analysed using CellQuest (BD Biosciences Pharmingen) software.
Anti-TLR neutralisation of cytokine production
Cells were incubated with anti-human TLR2 (clone TL2.1) or mouse IgG2a isotype control (20 μg·mL−1), for 30 min at room temperature or with anti-human TLR4 (clone HTA125) or mouse IgG2a isotype control (20 μg·mL−1) (all from eBioscience, CA, USA) for 1 h at 37°C. Thereafter, cells were stimulated with CSE (1.5%) and incubated overnight. Supernatants were collected and stored at -20°C prior to CXCL8 quantification.
Preparation of whole cell extracts
Neutrophils were plated at a density of 5×106 cells·mL−1 in 6-well cell culture plates and stimulated (as described previously) for 30 min. Cells were washed twice with PBS and lysed with lysis buffer containing 20 mM Tris pH 7.5, 1% Triton X-100, 100 mM NaCl, 40 mM NaF, 1 mM EDTA with protease inhibitors (MiniTM protease inhibitors; Roche Diagnostics, Burgess Hill, UK). Cells were subsequently lysed on ice for 5 min and, following centrifugation at 3,500×g for 5 min, the supernatants (whole cell extracts) were collected and frozen at -70°C.
Preparation of cytoplasmic and nuclear extracts
After activation, cells were washed twice with PBS and allowed to equilibrate for 5 min in ice-cold cytoplasmic extraction reagent (Pierce) containing protease inhibitors (MiniTM protease inhibitors, cocktail). Following centrifugation at 3,500×g for 5 min, the supernatants (cytoplasmic extracts) were collected and frozen at -70°C. The pellets were re-suspended in nuclear extraction buffer (Pierce) containing protease inhibitors. After vigorous mixing and incubating for 10 min on ice, the solution was centrifuged at 14,000×g for 5 min, and the supernatant (nuclear extract) was collected and stored at -70°C. Protein concentrations were determined by using a BCA protein assay kit.
Immunoprecipitation of endogenous IL-1R-associated kinase-1 and western blotting
Neutrophils (15×106) were stimulated with CSE (1.5%), CpG ODN (3 μM) for 10 min or pretreated with inhibitory ODN (10 mM) for 30 min and then stimulated with CSE (1.5%) for 10 min. After incubation for the indicated times, reactions were stopped with 5 mL of ice-cold saline with 2 mM phenylmethylsulfonylfluoride (PMSF) and rapid centrifugation. Then, the pellets were immediately frozen in dry ice after aspiration of the supernatants. Afterward, the pellets were lysed with 0.5 mL of ice-cold extraction buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 1% Triton X-100, 40 mM glycerophosphate, 1.5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, 2 mM dithiothreitol, 20 mM NaF, 2 mM sodium orthovanadate, 5 mM PMSF, 100 μg·mL−1 aprotinin, 10 μg·mL−1 leupeptin, 10 μg·mL−1 pepstatin. Following a 15-min incubation period on ice, samples were briefly vortexed, transferred to microtubes and centrifuged at 13,000×g, for 10 min at 4°C. Supernatants were collected and protein concentration was determined using the Micro BCA Protein Assay Reagent kit (Pierce) according to the manufacturer's instructions. Extracts with equal amounts of proteins were used for immunoprecipitation. The cell lysates (500 μg) were precleared by mixing with control rabbit IgG Abs (matched isotype) plus protein A-sepharose beads for 1 h at 4°C. Then, 4 μg of rabbit polyclonal IgG specific for IL-1R-associated kinase (IRAK)-1 was added to the precleared lysates and incubated at 4°C for 2 h on a rotator. Then, immune complexes were captured by the addition of 50 μl of prewashed protein A-Sepharose bead slurry and by incubation in a rotator for 1 h at 4°C. Sepharose beads were washed three times in ice-cold lysis buffer and then mixed 1:1 with 2× sample buffer and boiled for 5 min. Then, samples were subjected to SDS-PAGE, transferred to PVDF membrane and immunoblotted with rabbit polyclonal Ab anti-TRAF6 and detected with enzymatic chemiluminescence (ECL).
Western blotting
For western blot analysis, cells were washed once with cold PBS and lysed in ice-cold lysis buffer containing 50 mM Tris (pH 8.0), 110 mM NaCl, 5 mM EDTA, 1% Triton X-100 and 100 μg·mL−1 PMSF. Protein concentration was determined by BCA protein assay kit and (30–50 mg) protein was subjected were subjected to SDS-PAGE (10% (weight/volume)) gel. The separated proteins were electro-blotted on PVDF membranes. Membranes were then washed once with Tris-HCI pH 7.4, containing 159 mM NaCl and 1% Tween 20 (TBS-T), and then blocked in super-blocking buffer for 1 h. After washing with TBS-T, membranes were probed with antibodies against TLR9 and IRAK-1 and diluted of 1:3,000 in TBS-T and incubated overnight. After three washes with TBS-T, membranes were treated for 1 h with HRP-conjugated indicated antibodies diluted to 1:20,000 in TBS-T. After three washes with TBS-T, immunoreactive protein bands were revealed with an enhanced chemiluminescence western blot analysis system (ECL) or ECL Plus. Films were scanned and analysed on a GS7-10 Calibrated Imaging Densitometer equipped with Quantity One v. 4.0.3 software (Bio-Rad, Veenendaal, the Netherlands).
Quantification of NF-κB activity
NF-κB activation was detected using the Trans-AM NF-κB p65/NF-κB p50 Transcription Factor Assay Kit (Active Motif, Rixensart, Belgium) according to the manufacturer's instructions. Briefly, 2 μg nuclear extract was incubated with an oligonucleotide, containing the NF-κB consensus site, bound to a 96-well plate. After extensive washes, the NF-κB complexes bound to the oligonucleotide were incubated with an antibody directed against the NF-κB p65 subunit at a dilution 1:1,000. After washing, the plates were subsequently incubated with a secondary antibody conjugated to horseradish peroxidase (1:1,000), and the peroxidase reaction was quantified at 450 nm with a reference wavelength of 655 nm.
FACS analysis
Isolated neutrophils were incubated with CSE (1.5%) or CpG ODN (3 μM) alone or in combination at various time points and then fixed with formaldehyde (1%) and then permeabilised with permeabilisation buffer (eBioscience) and stained with PE-conjugated anti-human TLR9 Ab (eB72-1665) or a PE-conjugated rat IgG2a class-matched irrelevant Ab (eBioscience) as control for 30 min in permeabilisation buffer on ice. polymorphonuclear neutrophil (PMN), defined as CD16+CD3–CD19–HLA-DR– cells (all antibodies by ebioscience), were gated at a purity of >95.2%.
Cells were washed three times with immunofluorescence buffer (PBS 1%, FCS 1% and sodium azide 0.1%) and 10,000 cells were analysed on a FACSCalibur flow cytometer. The results obtained with specific antibodies were compared with those using isotype-matched control antibodies in parallel.
RNA isolation and real-time PCR
Total RNA for cDNA synthesis was prepared from activated and unactivated control cells at different time-points and then isolated using TRIzol reagent, and reverse transcription was performed in a 20-μl reaction with 1 μg total RNA, 50 mM Tris-HCl (pH 8), 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 500 μM each of deoxynucleotide triphosphates and SuperScript III reverse transcriptase at 42°C for 1 h. To eliminate DNA contamination, the RNA samples were incubated with DNase I at room temperature for 15 min. For real-time PCR, cDNA was analysed for the expression of TLR9 and β2-microglobulin (B2M) genes using Sybergreen Universal PCR Master Mix by using an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) at 50°C for 2 min, 95°C for 10 min, then 40 cycles of 95°C for 15 s and 60°C for 1 min. The sequences for PCR primers were: CXCL8: forward 5′-CTGGCCGTGGCTCTCTTG-3′ and reverse 5′-CCTTGGCAAAACTGCACCTT-3′, (accession number: NM_000584) ; TLR9: forward 5′-TGGTGTTGAAGGACAGTTCTCTC-3′ and reverse 5′-CACTCGGAGGTTTCCCAGC-3′ (accession number: NM_017442) ; and B2M: forward 5′-CTCCGTGGCCTTAGCTGTG-3′ and reverse 5′-TTTGGAGTACGCTGGATAGCCT-3′ (accession number: AF072097).
Stable transfection of NF-κB plasmid and NF-κB activity assay
TLR9 stably transfected HEK 293 cells were maintained in DMEM containing 10% FCS in 5% CO2 at 37°C. Cells were split on a 6-well dish or 60 mm dish at density 0.75×105cells·dish−1. After 18 h, cells were transfected with p-nifty 2×NF-kB SEAP plasmid using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. The total amount of DNA was adjusted using an empty vector, pcDNA3 (Invivogen). Cells were washed and cultured with 1 mg·mL−1 Zeocin for 4 weeks. At week 5 of transfection, cells were activated with CpG ODN (3 μM) and or CSE (1.5%) for 9 h for detection of SEAP by supernatant. For determination of SEAP as an indicator for NF-κB activity, the supernatants were subjected to QUANTI-BlueTM reagent as instructed by the manufacturer. SEAP levels were determined spectrophotometrically at 620–655 nm.
Statistical analysis
Experimental results are expressed as mean±sem. Results were tested statistically by an unpaired two-tailed t-test or one-way ANOVA, followed by Newman–Keuls test for comparing all pairs of groups. Analyses were performed by using GraphPad Prism (version 2.01). Results were considered statistically significant when p<0.05.
RESULTS
CSE and CpG-induces CXCL8 release via the generation of NO and ROS stress
CpG ODN induced the production of CXCL8 release by neutrophils in a concentration-dependent manner (fig. 1a). In all subsequent experiments, a sub-maximal concentration of 3 μM CpG ODN was selected. CSE (1.5%) also induced CXCL8 release from human neutrophils (fig. 1b and c). This effect on CXCL8 release was selective as CSE did not significantly induce the release of cytokines, such as TNF-α and IL-6, and chemokines, such as CCL2, CXCL10 and CCL11 (data not shown). Pre-incubation of the neutrophils with l-NAME or NAC (inhibitors of NO or ROS generation, respectively) inhibited the release of both CSE- and CpG ODN (3 μM) induced CXCL8 production back to baseline (fig. 1b and c). Pre-incubation of cells with the combination of sub-maximal concentrations of NAC and l-NAME further suppressed the CXCL8 production (fig. 1d) compared with that seen with either treatment alone, although the levels did not return to baseline.

Effects of NG-nitro-l-arginine methyl ester (l-NAME) and N-acetyl-cysteine (NAC) on the release of CXC chemokine ligand (CXCL)8 and generation of reactive oxygen species (ROS) and nitric oxide (NO) induced by cigarette smoke extract (CSE) and CpG oligonucleotides (ODN) of human neutrophils. Neutrophils (106 cells·mL−1) were seeded onto 96-well plates and placed in low-serum (1% fetal calf serum) medium and a) stimulated with various concentration of CpG ODN ( ) for 9 h (
) for 9 h ( : control); or left untreated (□) or pretreated with various concentrations of b) l-NAME (
: control); or left untreated (□) or pretreated with various concentrations of b) l-NAME ( ) or c) NAC (
) or c) NAC ( ) for 30 min and then stimulated with CSE (1.5%;
) for 30 min and then stimulated with CSE (1.5%;  ) or CpG ODN (3 μM;
) or CpG ODN (3 μM;  ) for 9 h. Levels of CXCL8 in supernatants were measured by ELISA and data are expressed as pg·mL−1. Assays were performed three times in duplicate. Data are presented as mean±sem (n = 3). a) ***: p≤0.001 compared with control. b and c) **: p≤0.01; ***: p≤0.001 compared with cells treated with l-NAME and NAC in activated cells with CSE and CpG ODN. d) Neutrophils were left untreated or pretreated with l-NAME (1 μM;
) for 9 h. Levels of CXCL8 in supernatants were measured by ELISA and data are expressed as pg·mL−1. Assays were performed three times in duplicate. Data are presented as mean±sem (n = 3). a) ***: p≤0.001 compared with control. b and c) **: p≤0.01; ***: p≤0.001 compared with cells treated with l-NAME and NAC in activated cells with CSE and CpG ODN. d) Neutrophils were left untreated or pretreated with l-NAME (1 μM;  ) or NAC (0.1 mM;
) or NAC (0.1 mM;  ) or in combination (
) or in combination ( ) for 30 min and then stimulated with CSE (1.5%;
) for 30 min and then stimulated with CSE (1.5%;  ) or CpG ODN (3 μM;
) or CpG ODN (3 μM;  ) or control ODN (5 μM) for 9 h. Levels of CXCL8 in supernatants were measured by ELISA and data are presented as mean±sem (n = 3). **: p≤0.01 compared with control; ##: p≤0.01 compared with CSE; ¶: p≤0.05 compared with CpG ODN; +: p≤0.05 compared to l-NAME alone or NAC alone. e–h) Neutrophils (106 cells) were incubated with CSE (1.5%;
) or control ODN (5 μM) for 9 h. Levels of CXCL8 in supernatants were measured by ELISA and data are presented as mean±sem (n = 3). **: p≤0.01 compared with control; ##: p≤0.01 compared with CSE; ¶: p≤0.05 compared with CpG ODN; +: p≤0.05 compared to l-NAME alone or NAC alone. e–h) Neutrophils (106 cells) were incubated with CSE (1.5%;  ) or CpG ODN (3 μM;
) or CpG ODN (3 μM;  ) for 5 h. Cells were washed and preincubated with e and f) 10 μM redox-sensitive dye DCFH-DA (for ROS) or g and h) 5 μM of diaminofluorescein diacetate (for NO) for 20 min at 37°C in PBS containing 1% BSA, in the dark. Generation of intracellular ROS and NO were determined by FACS analysis as described in the Material and Methods section and are representative of three independent experiments using polymorphonuclear neutrophil from different donors. The mean fluorescent intensity (MFI) of the groups is presented. Analyses of MFI from three independent experiments are presented as mean±sem in parts f and h for ROS and NO production, respectively. *: p≤0.05; **: p≤0.01 compared with control.
) for 5 h. Cells were washed and preincubated with e and f) 10 μM redox-sensitive dye DCFH-DA (for ROS) or g and h) 5 μM of diaminofluorescein diacetate (for NO) for 20 min at 37°C in PBS containing 1% BSA, in the dark. Generation of intracellular ROS and NO were determined by FACS analysis as described in the Material and Methods section and are representative of three independent experiments using polymorphonuclear neutrophil from different donors. The mean fluorescent intensity (MFI) of the groups is presented. Analyses of MFI from three independent experiments are presented as mean±sem in parts f and h for ROS and NO production, respectively. *: p≤0.05; **: p≤0.01 compared with control.
Furthermore, CSE (1.5%; fig. 1e and f) and CpG ODN (3 μM; fig. 1f and g) enhanced intracellular ROS and NO production.
CSE activates TLR9 signalling
To explore whether CSE regulates CXCL8 production via TLR9, neutrophils were pre-treated with chloroquine, an inhibitor of TLR activation pathways, particularly TLR9. Chloroquine (1–50 mg·mL−1) concentration-dependently attenuated the release of CXCL8 induced by CSE (fig. 2a). Chloroquine was cytotoxic at concentrations ≥10 mg·mL−1 (fig. 2b). Next, to examine specific activation of TLR9 signalling by CSE, the effects of the TLR9-inhibitory ODN on CXCL8 release was investigated 29. Inhibitory ODN suppressed the CSE-induced CXCL8 production in a concentration-dependent manner (fig. 2c). We then determined the effects of neutralising TLR2 and TLR4 antibodies on CXCL8 release by human neutrophils. As shown in figure 2d, a neutralising antibody against TLR4 significantly attenuated the release of CXCL8 by human neutrophils. A combination of chloroquine and the neutralising antibody against TLR4 further suppressed the release of CSE-induced CXCL8 production (fig. 2d).
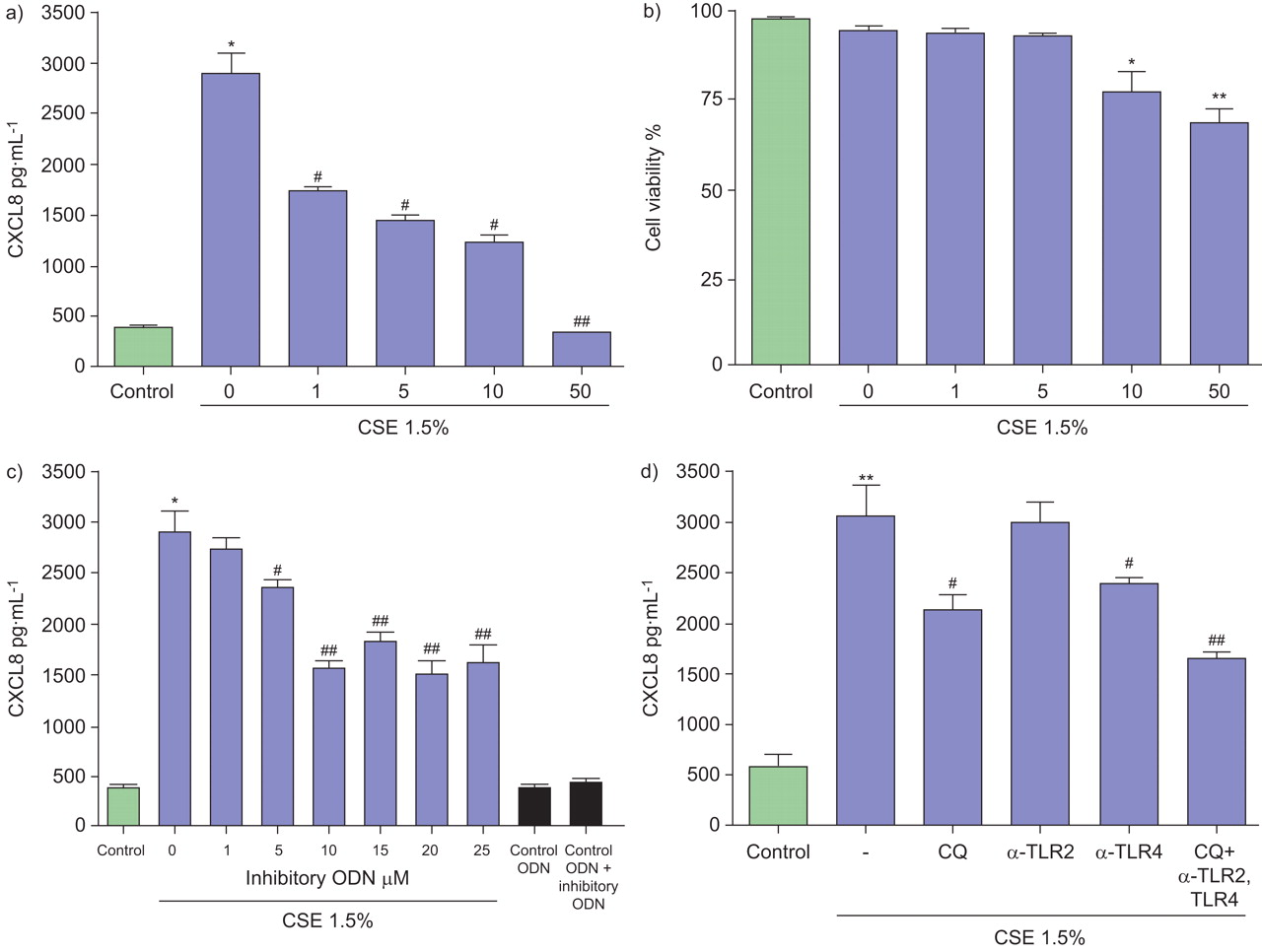
Effects of chloroquine (CQ), Toll-like receptor (TLR)9 inhibitory oligonucleotide (ODN) and blocking antibodies on CXC chemokine ligand (CXCL)8 release, induced by cigarette smoke extract (CSE) and CpG ODN of human neutrophils. Neutrophils (106 cells·mL−1) were seeded onto 96-well plates, placed in low-serum (1% fetal calf serum) medium and left untreated or pretreated with a and b) CQ (1–50 μg·mL−1), c) blocking inhibitory ODN (1–25 mM) or d) naturalising antibodies for TLR2 and TLR4 (20 μg·mL−1) with or without CQ for 30 min and then activated with CSE (1.5%;  ) or control ODN (5 mM) for 9 h. Levels of CXCL8 in culture supernatants were measured by ELISA and data are presented in pg·mL−1. Assays were performed three times in duplicate. b) Viability of the cells after incubation with various concentrations of CQ was determined by staining of cells with propidium ionide. Data are presented as mean±sem (n = 3). *: p≤0.05 compared with control; **: p≤0.01 compared with control; #: p≤0.05 compared with CSE; ##: p≤0.01 compared with CSE.
) or control ODN (5 mM) for 9 h. Levels of CXCL8 in culture supernatants were measured by ELISA and data are presented in pg·mL−1. Assays were performed three times in duplicate. b) Viability of the cells after incubation with various concentrations of CQ was determined by staining of cells with propidium ionide. Data are presented as mean±sem (n = 3). *: p≤0.05 compared with control; **: p≤0.01 compared with control; #: p≤0.05 compared with CSE; ##: p≤0.01 compared with CSE.
Further evidence for TLR9 involvement was suggested by the effects of CSE on TLR9 in a stably transfected HEK 293 cell line. CSE induced the release of CXCL8 in TLR9 stably transfected HEK 293 cells but not in TLR9 null cells (fig. 3a and b). CpG ODN served as a positive control for activation of TLR9 pathways (fig. 3). Components of cigarette smoke, such as nicotine and acrolein, may also modulate TLR9 signalling but we were unable to demonstrate any production of CXCL8 by these compounds in TLR9 stably transfected HEK 293 cells (data not shown).

Cigarette smoke extract (CSE) induces the production of CXC chemokine ligand (CXCL)8 of Toll-like receptor (TLR)9 stably transfected human embryonic kidney (HEK) 293 cell lines. a) TLR9 stably transfected HEK 293 and null HEK 293 cells (2×106 cells·mL−1) were stimulated with CSE (1.5%;  ) and CpG oligonucleotide (ODN; 3 μM;
) and CpG oligonucleotide (ODN; 3 μM;  ) for 9 h. CXCL8 levels in culture supernatants were measured by ELISA and data are presented as pg·mL−1. Assays were performed three times in duplicate. Data are presented as mean±sem (n = 3). b) TLR9 stably transfected HEK 293 or null cells stimulated for 6 h with CSE or CpG ODN and after lysis whole extracts subjected to western blot analysis with an anti-CXCL8 antibody or as a background with β-actin. The ratios of CXCL8 to β-actin expression from three separate gels are shown in the lower panels. Data are presented as mean±sem of triplicate samples. *: p<0.05; **: p≤0.01; ***: p≤0.001 compared with control.
) for 9 h. CXCL8 levels in culture supernatants were measured by ELISA and data are presented as pg·mL−1. Assays were performed three times in duplicate. Data are presented as mean±sem (n = 3). b) TLR9 stably transfected HEK 293 or null cells stimulated for 6 h with CSE or CpG ODN and after lysis whole extracts subjected to western blot analysis with an anti-CXCL8 antibody or as a background with β-actin. The ratios of CXCL8 to β-actin expression from three separate gels are shown in the lower panels. Data are presented as mean±sem of triplicate samples. *: p<0.05; **: p≤0.01; ***: p≤0.001 compared with control.
CSE modulates TLR9 expression in neutrophils
TLR9 is predominantly expressed in intracellular vesicles 17. Neutrophils were first fixed and permeabilised, and subsequently stained with a PE-conjugated anti-TLR9 Ab or isotype IgG control. After short exposure (5 h) to CSE, the intracellular expression of TLR9 was upregulated (fig. 4a) whereas after overnight exposure (24 h) TLR9 expression was downregulated (fig. 4b). Moreover, CSE did not induce surface expression of TLR9 (data not shown). This regulation of TLR9 protein expression paralleled with expression of mRNA (fig. 5a). Similar results were observed with CXCL8 mRNA expression (fig. 5b). Both NAC (1 mM) and l-NAME (0.1 μM) inhibited CSE and CpG ODN-enhanced intracellular TLR9 protein expression (fig. 5c).

Cigarette smoke extract (CSE) regulates Toll-like receptor (TLR)9 receptor expression. Neutrophils (106 cells·mL−1) were activated with CSE (1.5%) or CpG oligonucleotide (3 μM) for a) 5 h and b) 24 h. Then, cells were permeabilised and stained with an anti-human TLR9 antibody or an isotype control (immunoglobulin G) for 30 min in the dark. After two washes with cold PBS, the intracellular levels of TLR9 were determined by flow cytometry (using fluorescence-activated cells sorting). The results are representative for four experiments using neutrophils from different donors. Green lines indicated for control unstained cells, black lines indicated for control stained cells and blue lines indicated for cells treated with CSE. The mean fluorescent intensity (MFI) are presented.
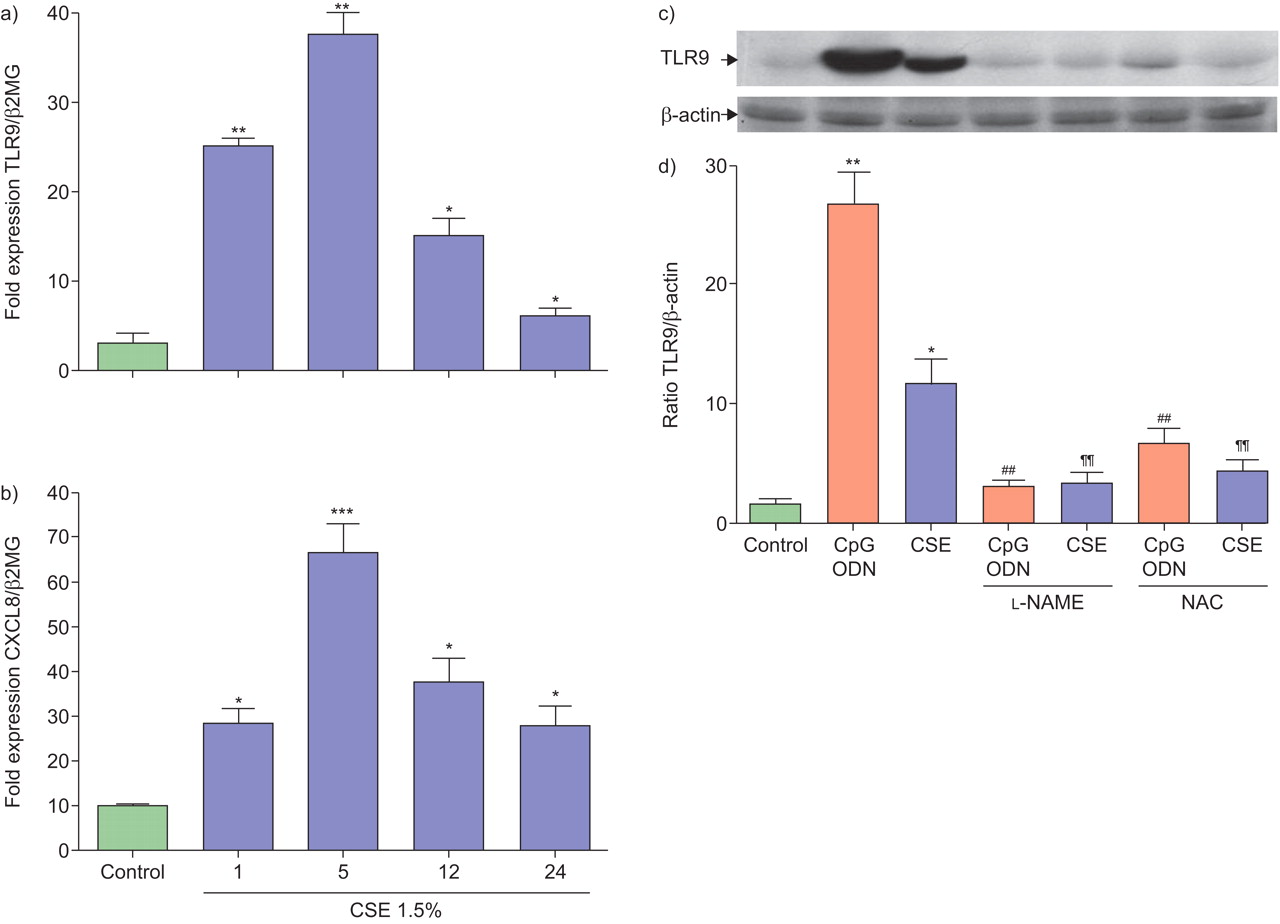
Cigarette smoke extract (CSE) modulates mRNA and intracellular expression levels of Toll-like receptor (TLR)9 and CXC chemokine ligand (CXCL) 8. Neutrophils (5×106 cells) were treated with CSE (1.5%;  ) for 1, 5, 12 and 24 h. Total RNA was isolated and after conversion to cDNA was subjected to real-time PCR analysis for mRNA expression for a) TLR9 or b) CXCL8, as a ratio of β2-macroglobulin expression (β2MG). Data are presented as mean±sem from three experiments. *: p<0.05, **: p≤001 compared with control. c) Neutrophils (5×106 cells) were activated with CSE (1.5%) or CpG oglionucleotide (ODN; 3 μM) for 5 h in the presence or absence of N-acetylcysteine (NAC; 1 mM) or NG-nitro-l-arginine methyl ester (l-NAME; 0.1 μM) and then lysed with lysis buffer. The expression of TLR9 and β-actin was detected using 50 mg of whole cell lysates by western blot analysis. Blots are representative of three independent experiments showing similar results. d) The ratio of TLR9 to β-actin expression from three separate gels is shown. Data are presented as mean±sem of triplicate samples. *: p<0.05; **: p≤0.01; ***: p≤0.001 compared with control; ##: p<0.01 compared with CpG ODN; ¶¶: p<0.01 compared with CSE alone.
) for 1, 5, 12 and 24 h. Total RNA was isolated and after conversion to cDNA was subjected to real-time PCR analysis for mRNA expression for a) TLR9 or b) CXCL8, as a ratio of β2-macroglobulin expression (β2MG). Data are presented as mean±sem from three experiments. *: p<0.05, **: p≤001 compared with control. c) Neutrophils (5×106 cells) were activated with CSE (1.5%) or CpG oglionucleotide (ODN; 3 μM) for 5 h in the presence or absence of N-acetylcysteine (NAC; 1 mM) or NG-nitro-l-arginine methyl ester (l-NAME; 0.1 μM) and then lysed with lysis buffer. The expression of TLR9 and β-actin was detected using 50 mg of whole cell lysates by western blot analysis. Blots are representative of three independent experiments showing similar results. d) The ratio of TLR9 to β-actin expression from three separate gels is shown. Data are presented as mean±sem of triplicate samples. *: p<0.05; **: p≤0.01; ***: p≤0.001 compared with control; ##: p<0.01 compared with CpG ODN; ¶¶: p<0.01 compared with CSE alone.
CSE induces activation of NF-κB via TLR9 signalling
In human neutrophils, CSE increased the activity of NF-κB in the nucleus and pretreatment with NAC, l-NAME and the TLR9 inhibitory ODN suppressed NF-κB activity induced by CSE (fig. 6a).

Cigarette smoke extract (CSE) activates Toll-like receptors and nuclear factor (NF)-κB pathways. Neutrophils (5×106 cells) were pretreated with inhibitory oligonucleotide (ODN; 10 μM) for 30 min and then stimulated with CSE (1.5%;  ) or CpG ODN (3 μM;
) or CpG ODN (3 μM;  ). a) Nuclear extracts (10 μg) were subjected to an electrophoretic mobility shift assay reaction for detection of NF-κB activity as described in the Materials and Methods section. Data are presented as mean±sem and are representative from one of five independent experiments. **: p<0.05, ***: p≤001 compared with control. #: p≤0.05 compared with CSE. b) Immunoblots of whole cell extracts obtained from neutrophils (15×106 cells) pretreated with inhibitory ODN (10 μM) and then stimulated with CSE (1.5%;
). a) Nuclear extracts (10 μg) were subjected to an electrophoretic mobility shift assay reaction for detection of NF-κB activity as described in the Materials and Methods section. Data are presented as mean±sem and are representative from one of five independent experiments. **: p<0.05, ***: p≤001 compared with control. #: p≤0.05 compared with CSE. b) Immunoblots of whole cell extracts obtained from neutrophils (15×106 cells) pretreated with inhibitory ODN (10 μM) and then stimulated with CSE (1.5%;  ) or CpG ODN (3 μM;
) or CpG ODN (3 μM;  ) for 30 min revealed with a rabbit polyclonal anti-interleukin-1R-associated kinase (IRAK)-1 antibody. Immunoblots are representative of at least three independent experiments. After stripping the blots, the membranes were incubated with β-actin as a housekeeping protein and visualised using enzymatic chemiluminescence. The ratio of IRAK-1 to β-actin expression from three separate gels is shown in the lower panels. Data are presented as mean±sem of triplicate samples. *: p<0.05 compared with control. d) Neutrophils (106 cells) were pretreated with inhibitory ODN and then stimulated with CSE or CpG ODN for 10 min. Cells were lysed and IRAK-1 was immunoprecipitated from the supernatants using antibodies specific for IRAK-1. Proteins were resolved using SDS-PAGE and analysed by western blot with anti-phosphotyrosine monoclonal antibody TRAF-6. OD: optical density; l-NAME: NG-nitro-l-arginine methyl ester; NAC: N-acetylcysteine.
) for 30 min revealed with a rabbit polyclonal anti-interleukin-1R-associated kinase (IRAK)-1 antibody. Immunoblots are representative of at least three independent experiments. After stripping the blots, the membranes were incubated with β-actin as a housekeeping protein and visualised using enzymatic chemiluminescence. The ratio of IRAK-1 to β-actin expression from three separate gels is shown in the lower panels. Data are presented as mean±sem of triplicate samples. *: p<0.05 compared with control. d) Neutrophils (106 cells) were pretreated with inhibitory ODN and then stimulated with CSE or CpG ODN for 10 min. Cells were lysed and IRAK-1 was immunoprecipitated from the supernatants using antibodies specific for IRAK-1. Proteins were resolved using SDS-PAGE and analysed by western blot with anti-phosphotyrosine monoclonal antibody TRAF-6. OD: optical density; l-NAME: NG-nitro-l-arginine methyl ester; NAC: N-acetylcysteine.
TLR activation triggers a signalling cascade that involves sequential recruitment and activation of IRAK-4 and -1 30. Phosphorylation of IRAK-1, in turn, enables recruitment and phosphorylation of the TAK1 complex which then dissociates, along with TRAF6, from the receptor complex. TRAF6 and IRAK-1 are ubiquitinated and degraded, enabling induction of TAK1 kinase activity and activation of NF-κB and the upregulation of proinflammatory gene transcription 30.
We, therefore, investigated whether CSE or CpG ODN affected IRAK-1 expression and the association of IRAK-1 with TRAF6. IRAK-1 underwent partial degradation upon stimulation with CSE and CpG ODN as evidenced in immunoblots of whole-cell extracts (fig. 6b). Pre-incubation of CSE-stimulated cells with the blocking inhibitory ODN abrogated IRAK-1 degradation. IRAK-1 was associated with TRAF6 in resting neutrophils (fig. 6c, lane 1). However, this association was lost following incubation of cells with CpG ODN and CSE for 10 min (fig. 6c, lanes 2 and 3, respectively) in agreement with the notion that IRAK becomes dissociated and degraded. The inhibitory ODN abrogated the dissociation and degradation of IRAK-1 (fig. 6c, compare lanes 3 and 4).
Next, as supportive evidence for CSE actions on the TLR9 pathway, the regulation of NF-κB in stably transfected dual TLR9/NF-κB reporter HEK 293 cells was investigated. The production of SEAP, an indicator of NF-κB activation in this system, was significantly enhanced after CSE incubation (fig. 7a). Similar results were obtained with CpG ODN as a positive control. The response was specific for TLR9 signalling since SEAP production was not increased by CSE or CpG in HEK 293 lacking TLR9 (fig. 7b).

Cigarette smoke extract (CSE) activates nuclear factor (NF)κB in Toll-like receptor (TLR)9 stably transfected human embryonic kidney (HEK) cells. TLR9, NFκB/secreted embryonic alkaline phosphatase (SEAP) double stably transfected HEK cells (a) and null, NFκB/SEAP stably transfectd HEK cells (b) were activated with CSE (1.5%;  ) or CpG oligonucleotide (ODN; 3 μM;
) or CpG oligonucleotide (ODN; 3 μM;  ) for 30 min. Levels of SEAP as a indicator for NF-κB activity in supernatants were measured by QUANTI-Blue™ reagent (Cayla-InvivoGen Europe, Toulouse, France). Assays were performed three times in duplicate. Data are presented as mean±sem (n = 3). *: p<0.05 compared with control.
) for 30 min. Levels of SEAP as a indicator for NF-κB activity in supernatants were measured by QUANTI-Blue™ reagent (Cayla-InvivoGen Europe, Toulouse, France). Assays were performed three times in duplicate. Data are presented as mean±sem (n = 3). *: p<0.05 compared with control.
DISCUSSION
The present study demonstrates that CSE induces the production of CXCL8 from human neutrophils via TLR9 signalling through a ROS- and NO-dependent mechanism. Chloroquine and the TLR9 inhibitory ODN attenuated the release of CXCL8-induced by CSE. Moreover, inhibitory ODN abrogated the induction of CXCL8 mRNA by CSE and inhibitory ODN also blocked TRAF6 and IRAK1 association and degradation and subsequent induction of NF-κB activity induced by CSE. Due to the short span of neutrophils in culture and their low transfection efficiency, we were not able to modulate TLR9 expression or signalling pathways in these cells. In support of our hypothesis, we report that CSE was able to induce CXCL8 release in HEK 293 cells stably transfected with TLR9 but not in cells devoid of TLR9. Internalisation and endosomal maturation have been shown to be required for CpG DNA to activate TLR9 signalling in immune cells 31, 32. Chloroquine, which effectively blocks endosomal maturation, significantly inhibited the CSE-induced increase of CXCL8 production in neutrophils, indicating a similar signalling pathway as CpG ODN.
In COPD patients, an increase of proinflammatory cytokines and chemokines including TNF-α and CXCL8 has been reported, and these mediators play an important role in establishing and maintaining the inflammatory condition, characterised by high local neutrophilia 7. Importantly, neutrophils constitutively express TLR9 33–35. TLRs have been intensely studied in the context of microbial challenges to inflammatory and immune cells, but their critical role in non-infectious challenges has only recently emerged. TLRs are the best characterised pattern recognition receptors in neutrophils in which lies most of their pathogen recognition capacity 33. Moreover, TLRs recognise distinct structural components of pathogens and trigger a signalling cascade that involves association of its intracellular Toll-IL-1R (TIR)-signalling domain with the adaptor molecule MyD88 36. A sequential recruitment and activation of IRAK-4 and -1 then occurs 37. Phosphorylation of IRAK-1, in turn, enables recruitment and phosphorylation of the TAK1 complex which then dissociates, along with TRAF6, from the receptor complex. TRAF6 and IRAK-1 are ubiquitinated and degraded enabling induction of TAK1 kinase activity and activation of NF-κB and the up-regulation of proinflammatory gene transcription 30. From this signalling complex, downstream cascades ultimately lead to activation of NF-κB, regulating proinflammatory gene transcription 30. TLRs are expressed in numerous cells within the airway and can therefore act as important sensors of environmental particulates and gases 23, 38, 39. TLRs have also been implicated in the pathogenesis and severity of autoimmune 40, cardiac and lung diseases 23, 38. In accordance with this concept, we and others 17, 23, 41, 42 have shown that cigarette smoke modulates TLR2 and TLR4 expression. In addition, CSE contains LPS 43 which is a potent activator of TLR4 signalling. Using a neutralising antibodies against TLR4 and TLR2, we have shown a partial involvement of TLR4 in CSE-induced CXCL8 expression in primary human neutrophils. Moreover, blocking TLR4 activity in combination with attenuation of TLR9 using chloroquine resulted in a greater decrease in CSE-induced CXCL8 expression. This indicates a degree of cross-talk between TLRs in regulating the response to cigarette smoke. The expression and coupling of receptors in different cell types will affect various signalling pathways, as well as that of NF-κB which may affect the final functional response observed. The fact that there is a residual level of CXCL8 production remaining after blocking TLR4 and TLR9 indicates the involvement of other pathways stimulated by CSE in the control of CXCL8 production.
Interestingly, we have found that CSE up-regulates TLR9 and CXCL8 expression in parallel. Moreover, the up-regulation of TLR9 expression was dependent on ROS and NO generation and reversed by longer-term incubation. The down-regulation of receptor expression by CSE is probably not due to cell toxicity, since the viability of the cells was not affected. Further investigations are required to understand these pathways and also the precise mechanisms that drive TLR activation.
TLR9, is to date the best-characterised sensor for bacterial DNA, containing short sequences of unmethylated CpG ODN motifs 38, and TLR9 stimulation results in alterations in cellular redox balance, peroxynitrite formation and activation NF-κB 44–49. Taken together, it can be concluded that CSE plays a crucial role in the induction of TLR9 expression and activation through generation of ROS and NO and the subsequent release of CXCL8 in neutrophils. TLR4 is also implicated in this effect and it is likely that other TLRs may also modulate CXCL8 production by CSE in an analogous manner.
Accumulating data indicated that cigarette smoke induces the production of free radicals in cells 47, 48. Free radicals in turn are able to activate a number of key pro-inflammatory transcription factors, including NF-κB and AP-1 49, 50. Indeed, CSE induced the cellular production of NO and ROS which could be protected by l-NAME and NAC. Inhibitory ODNs, that block TLR9 activation, attenuated CSE-induced NF-κB activity and upstream events in TLR9 signalling including the effects on TRAF and IRAK-1. Furthermore, CSE activated NF-κB in TLR9 stably transfected, but not untransfected, HEK 293 cells.
The precise component(s) of CSE that stimulate TLR9 and the mechanism by which it acts remains to be elucidated. It is possible that CSE contains CpG ODN which can directly stimulate TLR9 44, 46, 51. However, the differential effect of CSE and CpG ODN on ROS production suggests that this may not be the case. It has been suggested that ROS may activate TLRs either directly or indirectly 52, 53 potentially through an effect on modifying key residues within the TLR complex 54. Furthermore, future studies are needed to determine whether the ROS is derived directly from CSE or as a result of CSE activation of neutrophils.
Several studies have demonstrated that activation of TLR9 by CpG ODN could prevent allergic airway inflammation and airway hypersensitivity reaction, suggesting a potential therapeutic application of TLR9 signalling in asthma 55. In support of this, children who were exposed to parental smoking and those who took up cigarette smoking themselves, have a lower incidence of atopy to a range of common inhaled allergens 56. Intriguingly, in the current study we show that CSE increases the expression and activity of TLR9 in a temporal manner which may contribute to the prevention of allergic reactions or the resolution of inflammatory responses.
The role of the neutrophils in COPD is complex and it is unclear what the predominant driving force for their activation is in the human airway. Here we report that CSE directly stimulates neutrophil functions but we cannot determine what is the initial event that occurs in vivo? It is possible that inhaled cigarette smoke activates neutrophils recruited into the airways as a result of CXCL8 production by epithelial cells, for example, or that cigarette smoke depositing on the airway epithelium could be absorbed and activate circulating neutrophils which then migrate to the airways. The use of animal models will be able to address this in the future.
The observations and data presented here suggest a link between TLR9 activation and the release of CXCL8 by human neutrophils upon CSE exposure (fig. 8) which may contribute to the accumulation of neutrophils and inflammation within the airways of smokers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram of the induction of CXC ligand (CXCL)8 by human neutrophils via activation of Toll-like receptor (TLR)9 receptors by cigarette smoke. Cigarette smoke extract and CpG oligonucleotide (ODN) activate TLR9 pathway and generate reactive oxygen species (ROS) and NO production. These pathways may phosphorylate interleukin-1 receptor-associated kinase (IRAK) molecules. Following IRAK phosphorylation, the tumour necrosis factor receptor-associated factor 6 (TRAF6) adaptor protein interacts and induces translocation of the transcription factor nuclear factor (NF)-κB to the nucleus, resulting in transcriptional activation of genes encoding inflammatory mediators (e.g. CXCL8 and TLR9). N-acetylcysteine (NAC), NG-nitro-l-arginine methyl ester (l-NAME) and chloroquine act as blockers to the signalling.
Acknowledgments
This study was performed within the framework of Dutch Top Institute Pharma (project numbers D1.101). I.M. Adcock is supported by the Wellcome Trust.
Footnotes
Statement of interest
A statement of interest for this study is available from www.erj.ersjournals.com/site/misc/statements.xhtml
- Received April 14, 2009.
- Accepted April 9, 2010.
- ©ERS 2010