



Amifostine reduces lung vascular permeability via suppression of inflammatory signalling
- P. Fu 1 ,
- A. A. Birukova 1 ,
- J. Xing 1 ,
- S. Sammani 1 ,
- J. S. Murley 2 ,
- J. G. N. Garcia 1 ,
- D. J. Grdina 2 and
- K. G. Birukov 1
- 1Section of Pulmonary and Critical Care Medicine, Dept of Medicine, and 2Dept of Radiation and Cellular Oncology, University of Chicago, Chicago, IL, USA.
- K. G. Birukov, Section of Pulmonary and Critical Care Medicine, Dept of Medicine, University of Chicago, 929 E. 57th Street, GCIS Bldg, Room W410, Chicago, IL 60637, USA. Fax: 1 7738342683. E-mail: kbirukov{at}medicine.bsd.uchicago.edu
Abstract
Despite an encouraging outcome of antioxidant therapy in animal models of acute lung injury, effective antioxidant agents for clinical application remain to be developed. The present study investigated the effect of pre-treatment with amifostine, a thiol antioxidant compound, on lung endothelial barrier dysfunction induced by Gram-negative bacteria wall-lipopolysaccharide (LPS).
Endothelial permeability was monitored by changes in transendothelial electrical resistance. Cytoskeletal remodelling and reactive oxygen species (ROS) production was examined by immunofluorescence. Cell signalling was assessed by Western blot. Measurements of Evans blue extravasation, cell count and protein content in bronchoalveolar lavage fluid were used as in vivo parameters of lung vascular permeability.
Hydrogen peroxide, LPS and interleukin-6 caused cytoskeletal reorganisation and increased permeability in the pulmonary endothelial cells, reflecting endothelial barrier dysfunction. These disruptive effects were inhibited by pre-treatment with amifostine and linked to the amifostine-mediated abrogation of ROS production and redox-sensitive signalling cascades, including p38, extracellular signal regulated kinase 1/2, mitogen-activated protein kinases and the nuclear factor-κB pathway. In vivo, concurrent amifostine administration inhibited LPS-induced oxidative stress and p38 mitogen-activated protein kinase activation, which was associated with reduced vascular leak and neutrophil recruitment to the lungs.
The present study demonstrates, for the first time, protective effects of amifostine against lipopolysaccharide-induced lung vascular leak in vitro and in animal models of lipopolysaccharide-induced acute lung injury.
- Endothelium
- lipopolysaccharide
- lung
- permeability
- reactive oxygen species
Activation of tissue inflammation, oxidant-mediated tissue injury and increased vascular leak are cardinal features in the pathogenesis of acute lung injury (ALI) and the more severe acute respiratory distress syndrome (ARDS). In this condition, reactive oxygen and nitrogen species (ROS and RNS, respectively) produced by lung cells can oxidise and nitrate key lung proteins and phospholipids and inhibit their functions, or they may activate redox-sensitive pathological signalling pathways. Pro-inflammatory stimuli, such as bacterial wall-lipopolysaccharide (LPS), tumour necrosis factor (TNF)-α and interleukin (IL)-1, are potent triggers of lung inflammatory response. These same stimuli also elicit an oxidative cellular stress response 1, 2. An extensive body of evidence shows a crosstalk between the cellular signalling pathways and the cellular redox state through multiple mechanisms. For example, generation of ROS, such as H2O2, leads to activation of protein tyrosine kinases, mitogen-activated protein (MAP) kinases and their downstream effectors 3. However, ROS may also damage cells. At sites of inflammation and infection, the local cellular environment is enriched with ROS, cytokines and chemokines. Besides bactericide action, ROS can also attack and damage host tissues, and thus contribute to the pathogenesis of ARDS, reperfusion injury and other diseases. Levels of ROS/RNS correlate with the outcome of the disease and the severity of injury to the vascular endothelium and alveolar epithelium. Although the potential role of antioxidant enzymes and scavengers of ROS/RNS in reducing the severity of ALI has been recognised, the search for potential protective compounds continues. Animal and cell-culture models for testing potential therapeutic compounds in the ALI setting are yet to be developed.
Amifostine (S-2[3-aminopropylamino]ethylphosphorothioic acid; WR-2721) is a phosphorothioate that is converted to its active free thiol form by dephosphorylation by alkaline phosphatase in tissue. It has been approved by the Food and Drug Administration (FDA) for use as a cytoprotective agent to decrease the incidence of moderate-to-severe xerostomia in patients undergoing post-operative radiation therapy for the treatment of head and neck cancer 4. As a reducing agent capable of participating in intracellular reductive/oxidative processes, amifostine has the potential to affect redox-sensitive transcription factors and gene expression once inside the cell 5. Therefore, previous studies of amifostine focused on its radioprotective effects in tumour radiotherapy, where it acts as an oxidant scavenger. However, although recent studies have demonstrated a potential role of amifostine in pulmonary protection against bleomycin- or radiation-induced pulmonary toxicity 6, 7, it has not been tested yet as a potential protective therapy against sepsis and endotoxin-induced ALI. Compound WR-2721 is a stable but inactive precursor of WR-1065. When injected, WR-2721 is modified by membrane-bound alkaline phosphatase highly expressed in the endothelium, and transferred into the bioactive thiol metabolite WR-1065, which quickly penetrates into cell where the thiol groups act as free-radical scavengers and protect cells from oxidative damage. However, the endothelium is a major source of oxidants and may contribute to the oxidant-rich environment at the inflammatory locus.
The aim of the present study was to test the effects of the FDA approved compound amifostine against endothelial cell (EC) barrier dysfunction, induced by LPS and inflammatory cytokines, in vitro and to test its efficiency as pre-treatment in preventing the endotoxin-induced ALI in mouse model. The current results suggest that the protective effect of amifostine may be mediated by its antioxidant properties resulting in downregulation of oxidative stress and redox-sensitive signalling cascades, which lead to attenuation of lung vascular leak.
MATERIALS AND METHODS
Reagents and cell culture
Unless otherwise specified, all reagents including β-actin antibodies (catalogue No. A-5441) were obtained from Sigma (St Louis, MO, USA). The amifostine compounds WR-1065 (free thiol form used for cell-culture experiments) and WR-2721 (pro-drug formulation used in in vivo experiments) were obtained from the Drug Synthesis and Chemistry Branch, Division of Cancer Treatment, National Cancer Institute (Bethesda, MA, USA). IL-6 and IL-6 soluble receptor (SR) were obtained from R&D Systems (Minneapolis, MN, USA). VE-cadherin antibodies (catalogue No. sc-9989) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and nitrotyrosine antibodies (catalogue No. 32-1900) were purchased from Invitrogen (San Francisco, CA, USA). Antibodies against phosphorylated myosin light chain (MLC; catalogue No. 3674), p38 (catalogue No. 9216), heat shock protein (Hsp)27 (catalogue No. 2401), MAP kinase kinase (MEK)1/2 (catalogue No. 9121), extracellular signal regulated kinase (Erk)1/2 (catalogue No. 9101), nuclear factor (NF)-κB-p65 (catalogue No. 3031) and inhibitor (I)κBα (catalogue No. 9246) were obtained from Cell Signaling (Beverly, MA, USA). All reagents used for immunofluorescence staining were purchased from Molecular Probes, Inc. (Eugene, OR, USA). Human pulmonary artery endothelial cells (HPAEC) were obtained from Clonetics (Walkersville, MD, USA), cultured according to the manufacturer's protocol and used at passages 5–9.
EC imaging
EC monolayers grown on glass coverslips were stimulated with an agonist of interest, washed twice with room temperature PBS and fixed in 3.7% paraformaldehyde solution in PBS for 10 min at room temperature. This was followed by double immunofluorescence staining with Texas Red-conjugated phalloidin, to visualise actin filaments, and VE-cadherin antibody to visualise EC adherens junctions. EC cytoskeletal organisation was analysed as previously described 8.
Measurements of transendothelial electrical resistance
The cellular barrier properties were analysed by measurements of transendothelial electrical resistance across confluent endothelial cell monolayers using the electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY, USA). Cells were cultured on small gold electrodes (10−4 cm2), and culture media was used as an electrolyte. The total electrical resistance was measured dynamically across the monolayer using an electrical cell-substrate impedance sensing system (Applied Biophysics) and was determined by the combined resistance between the basal surface of the cell and the electrode, reflective of focal adhesion, and the resistance between the cells, as previously described 8.
Detection of ROS production in live cells
ROS production was measured using Image-iT LIVE Green Reactive Oxygen Species Detection Kit (Molecular Probes, Inc.). Confluent EC monolayers grown in 12-well plate were pre-treated with an unprotected form of amifostine for 30 min (WR-1065; 4 mM) or N-acetylcysteine for 1 h (NAC; 5 mM) followed by LPS stimulation for 6 h. After stimulation, cells were washed in 37°C Hanks buffer and incubated for 25 min with 1 mL of 25 µM carboxy-H2DCFDA working solution (37°C, protected from light). After washing three times in warm Hanks buffer, cells were subjected to microscopy using the Nikon video-imaging system (Nikon Inc., Tokyo, Japan) consisting of phase contrast inverted microscope equipped with a set of objectives and filters for immunofluorescence, and which was connected to a digital camera and image processor. In total, 10 fields were randomly chosen for each experiment condition. Immunofluorescence signal intensity was measured and expressed in arbitrary units per microscopic field.
Western blot analysis
Protein extracts from mouse lungs or EC lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes followed by incubation with specific antibodies of interest. Equal protein loading was verified by re-probing of membranes with anti-β-actin antibody. Immunoreactive proteins were detected using the enhanced chemiluminescent detection system according to the manufacturer’s protocol (Amersham Biosciences, Little Chalfont, UK). The relative intensities of immunoreactive protein bands (relative density units) were quantified by scanning densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA, USA).
In vivo model of ALI
Adult male, 8–10-week old C57BL/6J mice, with an average weight of 20–25 g (Jackson Laboratories, Bar Harbor, ME, USA), were anaesthetised with an intraperitoneal injection of ketamine (75 mg·kg−1) and acepromazine (1.5 mg·kg−1). LPS (0.7 mg·kg−1 body weight, Escherichia coli O55:B5, dissolved in sterile water) or sterile water was injected intratracheally in a small volume (20–30 µL) using a 20-gauge catheter (Penn-Century Inc., Philadelphia, PA, USA). Although water is a hypotonic solvent, it did not induce any noticeable injury in control animals injected with 20 μL sterile water, when compared with untreated mice or mice injected with 20 μL of normotonic physiological solution (data not shown). Mice were randomised to concurrently receive sterile saline solution or amifostine (WR-2721, 200 mg·kg−1) by intraperitoneal injection to yield the experimental groups: 1) control; 2) LPS (0.7 mg·kg−1) only; 3) WR-2721 (200 mg·kg−1) only; and 4) LPS (0.7 mg·kg−1) + WR-2721 (200 mg·kg−1). All animal experiments were approved by the University of Chicago Institutional Animal Care & Use Committee (Chicago, IL, USA) for the humane treatment of experimental animals.
Bronchoalveolar lavage fluid analysis
After 18 h, animals were sacrificed by exsanguination under anaesthesia. Tracheotomy was performed, and the trachea was cannulated with a 20-gauge i.v. catheter, which was tied into place. Bronchoalveolar lavage (BAL) was performed using 1 mL of warmed sterile Hanks balanced salt buffer (30°C). The collected BAL fluid was centrifuged at 520×g for 20 min at 4°C; the supernatant was removed and frozen at -80°C for subsequent protein study. The cell pellet was then resuspended in 1 mL of red blood cell-lysis buffer (ACK lysing buffer; Invitrogen) for 5 min. Red blood cell-lysis buffer was used to eliminate red cells from the cell pellet, as the presence of a large number of red cells in cell suspension may compromise the counting of white cells. White cells were then re-pelleted by centrifugation at 520×g for 20 min at 4°C. The cell pellet was again resuspended in 200 µL of PBS, and 20-µL aliquots were taken for cell counting using a hemocytometer. In brief, 20 µL of cell suspension was mixed with 20 µL of Trypan blue and allowed to sit at room temperature for 5 min. Cells were loaded onto a hemocytometer and then counted under a microscope. The remaining 180 µL of cell suspension were re-pelleted by centrifugation, and cell pellets were stored at -80°C for subsequent detection of myeloperoxidase (MPO) activity. The BAL protein concentration was determined using a Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA), which allowed the reaction to reach 90% of its maximum colour development within 15 min and colour changes of not more than 5% in 1 h. The absorbance was measured at 750 nm, and protein concentration was determined using standard curves.
Assessment of pulmonary vascular leakage by Evans blue
Evans blue dye (EBD, 30 mL·kg−1) was injected into the external jugular vein 2 h before termination of the experiment to assess vascular leak. In brief, at the end of the experiment, thoracotomy was performed and the lungs were perfused free of blood with PBS containing 5 mM EDTA. Left lung and right lungs were excised and imaged using a Kodak digital camera (Eastman Kodak Company, Rochester, NY, USA). After imaging, lungs were blotted dry, weighed, homogenised in PBS (1 mL·100 µg−1 tissue) and used for quantitative analysis of endobronchial tissue accumulation as described previously 9. Briefly, homogenised tissue was incubated with two volumes of formamide (18 h, 60°C) and centrifuged at 12,000 ×g for 20 min. Optical density of the supernatant was determined by spectrophotometry at 620 nm and 740 nm. EBD concentration (μg·g−1 tissue) in the lung tissue homogenates was calculated against a standard curve.
Histological assessment of lung injury
Left lungs were intratracheally instilled with 10% formalin from a height of 20 cm, immersed in 10% formalin for ≥24 h and then embedded in paraffin. After deparaffinisation and dehydration, the lungs were cut into 4-µm sections and stained with haematoxylin and eosin. Alveolar fluid accumulation and neutrophil infiltration as indices of lung leak and inflammation, respectively, were evaluated by bright field microscopy of lung tissue sections at ×40 magnification.
Statistical analysis
Results are presented as mean±sd from three to 10 independent experiments. Stimulated samples were compared to controls by unpaired t-test. For multiple-group comparisons, ANOVA, followed by the post hoc Fisher's test, was used. A p-value <0.05 was considered statistically significant.
RESULTS
Role of amifostine in the prevention of inflammatory agonist-induced hyperpermeability
To test the hypothesis that amifostine, a radiation protection compound with antioxidant properties, may attenuate lung endothelial hyperpermeability induced by inflammatory agents, the present authors examined the effects of amifostine on endothelial permeability induced by LPS and the inflammatory cytokine IL-6. EC treatment with H2O2 was used as an LPS-independent control for oxidative stress. Pre-treatment of HPAEC monolayers with a cell permeable free thiol form of amifostine, WR-1065 (0.4–4.0 mM), significantly attenuated H2O2-induced EC permeability judged by a marked decrease of transendothelial electrical resistance (fig. 1a⇓). The maximal protective effect of amifostine was achieved at a concentration of 4 mM. Similarly, amifostine alone did not affect basal transendothelial resistance (TER), but inhibited EC hyperpermeability induced by LPS (fig. 1b⇓). Amifostine at 4 mM is routinely used for in vitro studies and demonstrates cytoprotective effects without side-effects 10. For comparison, the NAC concentrations used in vitro were in the range of 10–20 mM 11, 12.

Effect of amifostine on H2O2-, lipopolysaccharide (LPS)- and interleukin (IL)-6-induced endothelial barrier dysfunction. Human pulmonary endothelial cells (EC) were grown on microelectrodes. At the timepoint indicated (grey arrow), cells were pre-treated with WR-1065 for 30 min. This was followed by stimulation with a) H2O2 (250 mM), b) LPS (200 ng·mL−1) or c) combination of IL-6 (25 ng·mL−1) and its soluble receptor (SR; 100 ng·mL−1). Transendothelial resistance reflecting EC permeability changes was monitored over time. Pooled data from three independent experiments are shown. control: black; H2O2 250 μM: pink; amifostine 4 mM+H2O2: orange; amifostine 1 mM+H2O2: green; amifostine 0.4 mM+H2O2: pale blue; LPS 200 ng·mL−1: grey; amifostine 4 mM+LPS: red; amifostine 4 mM: yellow; IL-6+SR: purple; amifostine 4 mM+(IL-6+SR): dark blue.
Activation of IL-6 production in the lungs induced by LPS intratracheal instillation has been previously shown 8. In vitro, a combination of IL-6 and its SR increases pulmonary EC permeability 13. Pre-treatment of EC with amifostine significantly reduced TER increases in response to IL-6+SR challenge (fig. 1c⇑). Taken together, these data strongly suggest a protective effect of amifostine pre-treatment against EC barrier dysfunction induced by inflammatory agonists.
Effects of amifostine on endothelial cytoskeletal remodelling and disruption of adherens junctions induced by inflammatory agonists
In the following experiments, the present authors assessed the effects of amifostine on cytoskeletal remodelling and disruption of adherens junctions caused by H2O2, LPS and IL-6 with its soluble receptor (IL-6+SR). After agonist challenge, cytoskeletal remodelling and integrity of adherens junctions in control and amifostine-pre-treated pulmonary EC were examined by immunostaining for F-actin and VE-cadherin. In unstimulated cells and cells treated with amifostine alone, F-actin was primarily organised into actin bundles randomly distributed in the cell (fig. 2a–h⇓). Upon stimulation with H2O2, LPS, or IL-6 + SR, F-actin was reorganised into thicker stress fibres in the centre of the cells. These changes were associated with appearance of paracellular gaps (shown by arrows) indicating EC barrier compromise. Remarkably, amifostine pre-treatment attenuated agonist-induced stress fibres and gap formation. In agreement with these data, pre-incubation of pulmonary EC with amifostine also attenuated H2O2- and LPS-induced disruption of monolayer integrity, as detected by immunofluorescence staining for VE-cadherin (fig. 2⇓i–n). These results suggest that the protective role of amifostine may be attributed to its ability to attenuate cytoskeletal remodelling induced by H2O2, LPS and IL-6. These results show that protective effects of amifostine against H2O2-, LPS- or IL-6-induced hyperpermeability are also associated with the preservation of cytoskeletal and cell–cell junction organisation.

Amifostine (Amif) prevents agonist-induced lung endothelial cell (EC) cytoskeletal remodelling and adherens junction disruption. EC grown on glass coverslips were pre-treated with Amif 4 mM for 30 min or vehicle followed by stimulation with H2O2 for 15 min (250 mM), lipopolysaccharide (LPS) for 6 h (200 ng·mL−1), or interleukin (IL)-6+soluble receptor for 6 h (25 ng·mL−1 and 100 ng·mL−1). a–h) Actin cytoskeletal remodelling was assessed by immunofluorescence staining with Texas Red phalloidin. The paracellular gap formation is indicated by arrows. a–d) Vehicle, H2O2, LPS and IL-6, respectively. e–h) Amif, Amif +H2O2, Amif +LPS and Amif+IL-6, respectively. i–n) VE-cadherin staining was performed to visualise adherens junctions. LPS-induced disruption of adherens junctions is indicated by arrows. i–k) Vehicle, H2O2 and LPS, respectively. l–n) Amif, Amif +H2O2 and Amif +LPS, respectively. The panels are representative of the entire cell monolayer. Results of three independent experiments are shown.
Effects of amifostine on ROS production induced by LPS
To test the hypothesis that amifostine may attenuate LPS-induced EC cytoskeletal remodelling and barrier dysfunction by its ability to scavenge ROS, ROS production in the EC stimulated with LPS was measured, with or without amifostine pre-treatment. An antioxidant NAC was used as a positive control. LPS treatment for 6 h increased ROS production in pulmonary EC in a dose–dependent manner with maximal effect at 500 ng·mL−1 LPS (fig. 3⇓). Pre-treatment with amifostine or NAC significantly reduced ROS production by LPS. Importantly, amifostine was more effective in suppression of LPS-induced ROS production. These results suggest that the protective role of amifostine in preventing H2O2-, LPS- or IL-6+SR-induced EC barrier dysfunction may be mediated, at least in part, by its antioxidant activity.
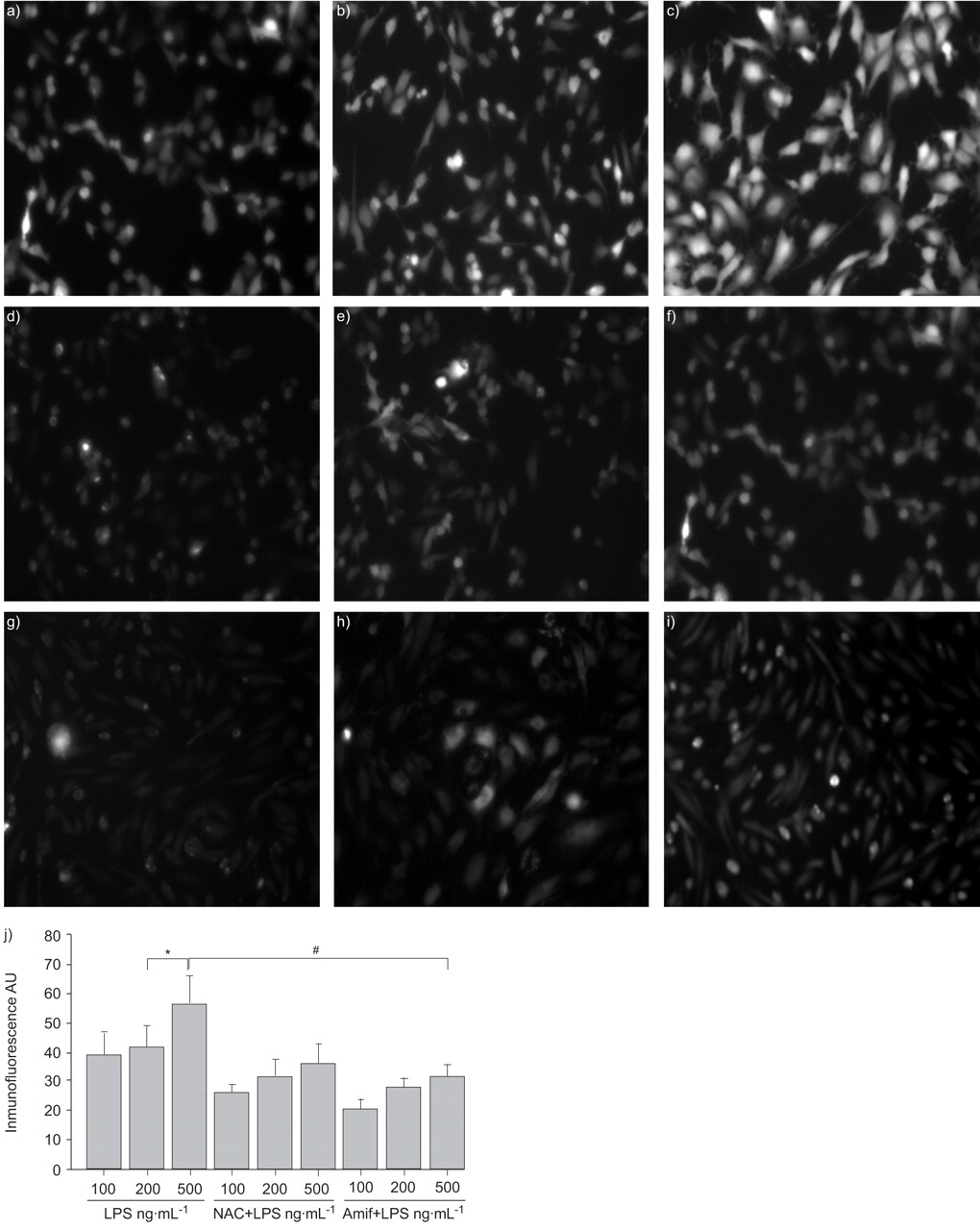
Amifostine (Amif) inhibits reactive oxygen species (ROS) production induced by lipopolysaccharide (LPS). Human pulmonary artery endothelial cells were pre-treated with a–c) vehicle, d–f) N-acetylcysteine (NAC; 5 mM, 1 h), or g–i) WR-1065 (4 mM, 30 min). This was followed by 6 h LPS stimulation of a, d and g) 100 ng·mL−1, b, e and h) 200 ng·mL−1, or c, f and i) 500 ng·mL−1. ROS was detected by carboxy-H2DCFDA and carboxy-H2DCFDA-5-(and 6)-carboxy-2′,7′-dichlorodihydrofluorescin diacetate fluorescent assay, as described in the Materials and Methods. The results are representative of the entire cell monolayer and have been reproduced in three independent experiments. j) Statistical analysis of the effect of NAC and Amif on LPS-induced ROS production. AU: arbitrary units. *: p<0.05, compared with 500 ng·mL−1 LPS treated endothelial cells; #: p<0.005, compared with NAC or amifostine pre-treated EC.
Regulation of oxidative stress-sensitive signalling molecules by amifostine
ROS and associated oxidative stresses are shown to activate various signalling molecules, such as Erk1/2, p38, JNK MAP kinases and NF-κB signalling, which are intimately involved in the control of EC permeability and inflammation and regulated by phosphorylation on serine/threonine and tyrosine residues.
In the next series of experiments, the present authors investigated the effects of amifostine on the regulation of MAP kinase- and NF-κB-dependent signalling activated by oxidative stress. Western blot analysis of pulmonary EC stimulated with or without amifostine pre-treatment showed that amifostine abolished LPS-, IL-6- and H2O2-induced activation of Erk1/2 and p38 MAP kinase cascades (fig. 4⇓), as detected by phosphorylation of MEK1/2, Erk1/2, p38 MAP kinases and the p38-dependent regulator of actin dynamics, Hsp27. Amifostine dramatically attenuated LPS-induced phosphorylation of IκBα and the NF-κB-p65 subunit (fig. 4a⇓), which are critical for activation of NF-κB-dependent transcription. Similar results were obtained in EC monolayers stimulated with H2O2 (fig. 4b⇓).

Amifostine (Amif) attenuates intracellular signalling activated by inflammatory agonists. Human pulmonary artery endothelial cells were pre-treated with Amif (4 mM, 30 min) or vehicle followed by stimulation with a) lipopolysaccharide (LPS; 200 ng·mL−1, 2 h), b) H2O2 (250 mM, 15 min), or c) interleukin (IL)-6 + soluble receptor (25 ng·mL−1 and 100 ng·mL−1, 2 h). Phosphorylation (p) of p38, mitogen-activated protein kinase kinase (MEK)1/2, extracellular signal regulated kinase (Erk1/2), inhibitor (I)κBα, nuclear factor (NF)-κB-p65, myosin light chain (MLC) and heat shock protein (Hsp27) was detected with corresponding phospho-specific antibodies. Results are representative of three independent experiments.
Increased MLC phosphorylation is associated with increased actin polymerisation and activation of actomyosin contraction. Additionally, inflammatory mediators may inactivate myosin-associated protein phosphatase 14 and thus further promote MLC phosphorylation, a marker of endothelial cell contraction. In the following experiments the current authors analysed the effects of amifostine on phosphorylation of MLC induced by LPS, IL-6 or H2O2. Pre-treatment with amifostine significantly inhibited phosphorylation of MLC induced by these agonists (fig. 4c⇑), which is consistent with protective effects of amifostine on the EC barrier under oxidative stress and inflammatory stimulation.
Taken together, the results of the cell-culture experiments strongly suggest that the activation of p38, Erk1/2 MAP kinase and NF-κB pathways are associated with EC stress responses to inflammatory agonists, and indicate a potent protective effect of amifostine against EC oxidative stress, barrier dysfunction and cytoskeletal remodelling.
Effects of amifostine on LPS-induced cell infiltration in mouse lungs
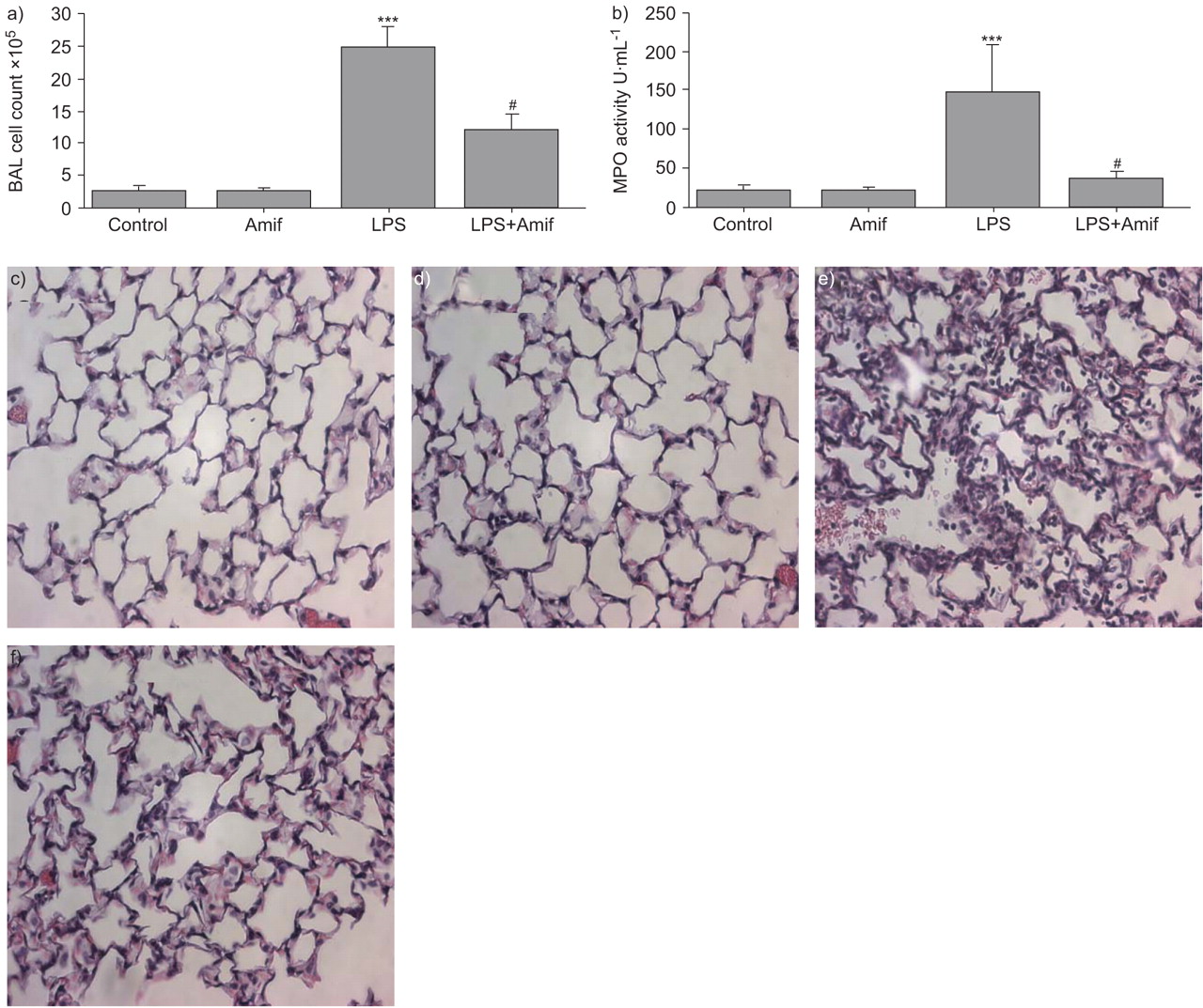
To evaluate a role of amifostine in the modulation of lung permeability, an in vivo model of LPS-induced ALI was used. The pro-drug form of amifostine was used in the in vivo studies. LPS induced dramatic lung injury with a nearly 10-fold increase in BAL cell counts at 18 h (2.52±0.76×105 cells·mL−1 versus 2.46±0.33×106 cells·mL−1, p<0.001; fig. 5a⇓). The influx of neutrophils into the lungs in response to LPS was inhibited by treatment with amifostine (2.46±0.33×106 cells·mL−1 versus 1.22±0.24×106 cells·mL−1, p<0.001). Correspondingly, the activity of MPO, a marker of tissue oxidative stress and inflammation induced by activated neutrophils, was dramatically decreased by amifostine pre-treatment, as compared to LPS alone (37±8.5 U·mL−1 versus 148.3±61.4 U·mL−1, p<0.01; fig. 5b⇓).

Amifostine (Amif) attenuates lipopolysaccharide (LPS)-induced neutrophil infiltration increased myeloperoxidase (MPO) activity. C57BL/6J mice were treated with LPS (0.7 mg·kg−1), with or without concurrent treatment with WR-2721 (200 mg·kg−1) for 18 h. Control animals were treated with sterile water or WR-2721 (200 mg·kg−1) alone. After stimulation, the a) bronchoalveolar lavage (BAL) cell count and b) MPO activity were measured in BAL fluid taken from control and experimental animals. n = 6–9 per group. c–f) Control, Amif, LPS and LPS+Amif, respectively. Lungs were excised, fixed in 4% paraformaldehyde, embedded in paraffin and used for histochemical analysis after haematoxylin and eosin staining. Images are representative of 6–9 lung specimens for each condition. ***: p<0.001 versus control; #: p<0.001 versus LPS.
Histological analysis of paraffin-embedded lung tissue sections stained with haematoxylin and eosin was performed to further examine the effects of amifostine on LPS-induced changes in lung microstructure. LPS stimulation for 18 h induced significant neutrophil accumulation in the lung parenchyma, which was significantly reduced by amifostine treatment (fig. 5c–f⇑). These results clearly demonstrate that amifostine may inhibit LPS-induced ALI.
Effects of amifostine on LPS-induced lung vascular leak
In the following studies, the effects of amifostine on LPS-induced lung vascular leak were examined. Analysis of BAL protein content was used as an index of lung vascular permeability. Intratracheal instillation of LPS significantly increased in the total protein concentration in BAL fluid (fig. 6a⇓), as compared with control animals (0.14±0.04 mg·mL−1 versus 1.01±0.13 mg·mL−1, p<0.001). Concurrent treatment with LPS and amifostine significantly attenuated BAL protein concentrations, in comparison to LPS alone (0.46±0.12 mg·mL−1 versus 1.01±0.13 mg·mL−1, p<0.001). There was no significant difference in BAL protein in amifostine-treated animals and controls in the absence of LPS.
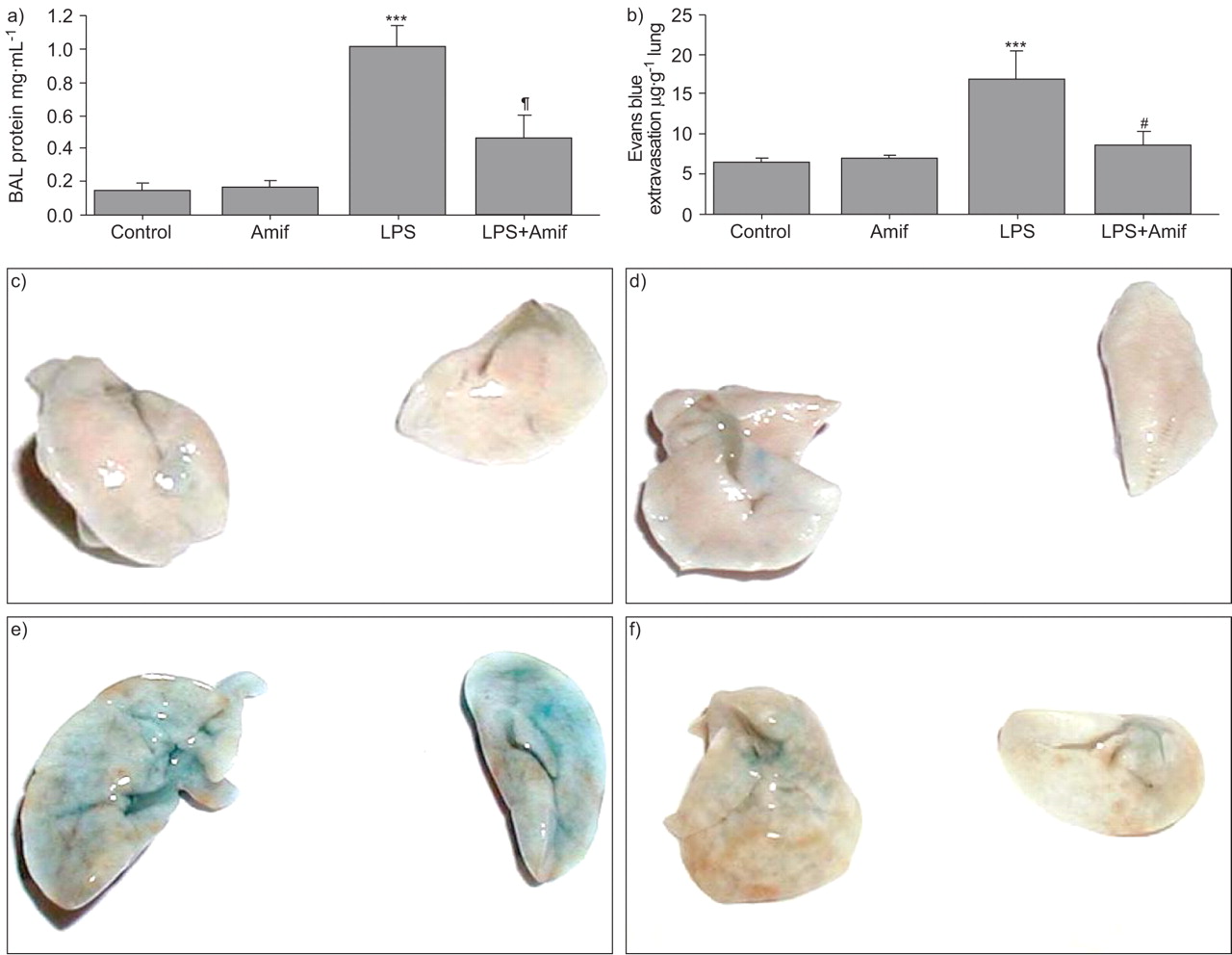
Amifostine (Amif) attenuates lipopolysaccharide (LPS)-induced lung vascular leak. C57BL/6J mice were treated with LPS (0.7 mg·kg−1), with or without concurrent treatment with WR-2721 (200 mg·kg−1) for 18 h. Control animals were treated with sterile water or WR-2721 (200 mg·kg−1) alone. a) Protein concentration was measured in bronchoalveolar lavage fluid taken from control and experimental animals. b) Lung vascular permeability was assessed by Evans blue accumulation in the lungs as described in the Materials and Methods. n = 3 per group. c–f) Control, Amif, LPS and LPS+Amif, respectively. Images of the lungs represent Evans blue leakage into lung tissue. Results are representative of three independent experiments. Quantitative spectrophotomertic analysis of Evans blue-labelled albumin extravasation. ***: p<0.001 versus control; #: p<0.001 versus LPS.
The protective effects of amifostine against LPS-induced lung vascular barrier compromise were further assessed by measurement of Evans blue leakage into the lung tissue. LPS caused substantial Evans blue leakage from the vascular space into the lung parenchyma, which was significantly decreased by amifostine treatment (fig. 6c–f⇑). These results were confirmed by quantitative analysis of Evans blue-labelled albumin extravasation in the lung preparations (fig. 6b⇑). Evans blue accumulation in the lungs from LPS-treated mice was significantly increased (16.95±3.46 μg·g−1 wet weight lung versus 6.63±0.40 μg·g−1 wet weight lung in non-treated controls, p<0.001). LPS-induced tissue accumulation of Evans blue was reduced by amifostine treatment (8.77±1.48 μg·g−1 wet weight lung versus 16.95±3.46 μg·g−1 wet weight lung, p<0.001). Thus, the data demonstrate protective effects of amifostine in the mouse model of vascular permeability induced by LPS.
Effects of amifostine on LPS-induced oxidative stress-sensitive signalling in vivo
The present authors then examined whether the protective effect of amifostine in vivo was associated with its antioxidant properties. LPS-induced ROS production, nitric oxide (NO) synthase activation and increased NO generation lead to rapid reaction of NO with superoxide and generation of peroxynitrite 15, leading to nitration of protein tyrosine residues. Thus, the presence of proteins with nitrated tyrosine residues can be used as a marker of oxidative stress in vivo, including in ARDS 16. Western blot analysis of lung tissue samples from LPS- and amifostine+LPS-treated mice was performed to detect the presence of tyrosine nitrated proteins. LPS treatment increased the content of nitrotyrosine-containing proteins in the lung tissue samples with major bands observed in the 25 kDa and 50 kDa range, which was significantly attenuated by amifostine treatment (fig. 7a⇓, upper panel). After 6 h of LPS challenge, nitrotyrosine-containing proteins were mainly detected in the 50 kDa range. Amifostine pre-treatment significantly decreased LPS-induced protein nitration. In addition, amifostine dramatically attenuated LPS-induced phosphorylation of p38 MAP kinase (fig. 7a⇓, middle panel). These results strongly suggest that the protective effect of amifostine in vivo is associated with the reduction of LPS-induced oxidative stress and redox-sensitive stress kinase signalling.

Amifostine (Amif) inhibits lipopolysaccharide (LPS)-induced oxidative stress-sensitive signalling in mouse lungs. a and b) C57BL/6J mice were treated with LPS (0.7 mg·kg−1), with or without concurrent treatment with WR-2721 (200 mg·kg−1). Control animals were treated with sterile water or WR-2721 (200 mg·kg−1) alone. Mice lungs were harvested after 2 or 6 h of LPS instillation, and tissue samples were prepared for western blot analysis. Accumulation of nitrotyrosinated proteins and p38 mitogen-activated protein kinase phosphorylation was detected using specific antibodies. Equal protein loading was confirmed by re-probing of membranes with β-actin antibodies. Results are representative of three independent experiments. #: p<0.005. c and d) Dose dependence of Amif effects on LPS-induced neutrophil accumulation and protein content in bronchoalveolar lavage (BAL). Mice were treated with LPS (0.7 mg·kg−1), with or without concurrent treatment with the range of WR-2721 doses (25–200 mg·kg−1). After 18 h of stimulation, c) cell count and d) protein concentration were measured in BAL fluid taken from control and experimental animals. n = 6–9 per group. RDU: relative density units. ***: p<0.001 versus control; #: p<0.001 100 mg·kg−1 versus 200 mg·kg−1 WR-2721.
The following experiments tested a dose dependence of amifostine protective effects in the LPS model of lung vascular leak. Amifostine exhibited protective effects against LPS-induced increases in the BAL cell count (fig. 7c⇑) and protein content (fig. 7d⇑) in the 25–200 mg·kg−1 dose range. Remarkably, even low doses of amifostine (25 mg·kg−1) exhibited a nearly maximal protective effect against BAL protein accumulation, although maximal suppression of BAL cell accumulation was achieved at dose of 200 mg·kg−1 amifostine.
DISCUSSION
Although ROS generated under physiological conditions play an important role in cell homeostasis and cell signal transduction 17, their excessive generation at sites of inflammation becomes detrimental and may cause tissue injury. The vascular endothelium, which regulates the passage of macromolecules and circulating cells from blood to tissue, becomes readily exposed to oxidative stress and plays a critical role in the pathophysiology of several vascular disorders. The present study demonstrated that amifostine, which acts as a donor of active free thiol, inhibited LPS-induced endothelial monolayer disruption and attenuated LPS-induced lung vascular leak via downregulation of ROS and tissue oxidative stress.
LPS is an important trigger of the lung inflammation involved in the pathogenesis of lung oedema and ARDS. Through binding to its receptor, CD14, on the cell membrane, LPS induces the release of pro-inflammatory cytokines. As a result of LPS challenge, activated neutrophils migrate into the lung interstitium from the blood circulation and produce significant amounts of ROS leading to further escalation of inflammation. Activated endothelial cells may be another important source of ROS contributing to the formation of an oxidant-rich environment at the sites of inflammation. The current results show significant ROS production by pulmonary EC induced by LPS. Furthermore, H2O2-, LPS- and IL-6-induced stress fibre formation and disruption of adherens junctions in pulmonary EC monolayers was prevented by amifostine. The barrier protective effect of amifostine observed in the present study was linked to its ability to inhibit ROS production and attenuate redox-dependent pro-inflammatory signalling in the EC cultures and in the murine model of LPS-induced ALI.
NF-κB is a redox-sensitive transcription factor activated upon LPS stimulation, which triggers inflammatory signalling and activates expression of pro-inflammatory cytokines. The current data show that potent inhibitory effects of amifostine pre-treatment on the LPS-induced NF-κB activation. Erk1/2 and p38/MK-2 MAP kinase pathways and NF-κB may become activated in a redox-sensitive manner 3. In addition, p38 MAP kinase may be involved in actin filament reorganisation in response to oxidative stress via MAP kinase-dependent phosphorylation of the actin-binding effector Hsp27. The present study shows that phosphorylation of p38 and Hsp27 caused by LPS or H2O2 was attenuated by amifostine in vitro and in vivo. In addition, amifostine pre-treatment abolished LPS-, H2O2- and IL-6-induced activation of regulatory MLC, which triggers EC contraction and barrier dysfunction. Inflammatory cytokine IL-6 is a marker of inflammation elevated in the model of endotoxin-induced lung injury 8, 18. Soluble IL-6 receptor (SR) is a 50–55 kDa ligand binding protein, which can bind its ligand and induce cellular responses by association with gp130, thus acting as an IL-6 agonist. The association of IL-6 with the soluble form of SR-α is capable of eliciting a biological response in cells that only express the membrane gp130. It has previously been described that single treatment of HPAEC with IL-6 or its SR did not significantly change basal TER levels, whereas a combination of IL-6 and SR induced a significant TER decrease 13. In the current study, a combination of IL-6 and its SR treatment was used to reproduce inflammatory reactions in pulmonary endothelium. In these experiments, IL6/SR-induced EC barrier dysfunction, cytoskeletal remodelling and phosphorylation of the p38 MAP kinase target, Hsp27, was attenuated by amifostine (figs 1⇑, 2⇑ and 4⇑). Collectively, these results demonstrate that the LPS-induced oxidative stress plays an essential role in the activation of inflammatory signalling and cytoskeletal remodelling leading to EC permeability and lung vascular leak, which can be markedly attenuated by amifostine pre-treatment.
Western blot detection of nitrotyrosinated proteins, a footprint of oxidative stress, showed increased lung tissues levels of nitrotyrosinated protein immunoreactivity after 2 and 6 h of LPS exposure, which were markedly attenuated by amifostine (fig. 8⇓). Antioxidant strategies for attenuation of lung inflammatory injury have been tested in previous studies. For example, NAC, a thiol antioxidant compound, exhibited antioxidant effects and suppressed ROS-mediated lung injury 20. Similar to amifostine, NAC attenuated activation of p38 and MAP kinase kinase (MKK)3/MKK6 in the TNF-α model of EC barrier dysfunction 21. However, NAC application in clinical settings did not prove its efficiency. In fibrous alveolitis, where activated inflammatory cells induce oxidative stress in a lower respiratory tract, high doses of NAC (1.8 g daily for 12 weeks in addition to immunosuppressive therapy) did not significantly suppress inflammatory cell activation 22. A Nordic multicentre controlled trial 23 showed that a 6-day course of i.v. NAC during the first week of life does not prevent bronchopulmonary dysplasia or death, or improve lung function at term in infants with extremely low birth weight. Although previous studies demonstrated protective effects of amifostine on radiation (or chemotherapy) induced acute lung toxicity, there is no direct evidence of an amifostine protective effect in the model of endotoxin-induced ALI. The current results indicate potent protective effects of amifostine against LPS-induced pulmonary EC barrier compromise in vitro and LPS-induced lung leukocyte infiltration, BAL protein accumulation and vascular leak. Amifostine inhibited LPS-induced leukocyte accumulation in BAL in a dose–dependent manner. A maximum protective effect was achieved at 200 mg·kg−1. However, marked protective effects were still observed at significantly lower dose (25 mg·kg−1), which do not cause toxic side-effects. A dose of 910 mg·m−2 amifostine was originally established for use in clinical chemotherapy studies. At this concentration, side-effects including nausea, vomiting, hypotension and hypocalcaemia were recorded. In the current study, the protective effects of amifostine against lung injury were observed even at substantially lower doses of amifostine without introducing side-effects. These results suggest a potential use of amifostine as clinical treatment of ALI syndromes. Another advantage of amifostine over other antioxidants, including NAC, is comparable efficiency at different routes of administration. For the most antioxidant agents, the maximal effect is usually achieved by intravenous injection. In contrast, amifostine exhibits comparable effects during subcutaneous and intravenous 24, as well as intraperitoneal 25 administration.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of lipopolysaccharide (LPS)-induced inflammation and endothelial barrier dysfunction by amifostine. Lung exposure to LPS results in activation of Toll-like receptor (TLR)4 via LPS binding with LPS binding protein (LBP) and phosphatidylinositol glycan-linked cell surface protein, CD14. Subsequently, the LPS-CD14 complex interacts with TLR4 and its accessory protein MD2. An alternative pathway of LPS-induced signalling may involve caveolae 19. LPS-signalling complex is assembled in caveolae, and the intracellular adaptor proteins MyD88 and MAL are recruited to the Toll/interleukin (IL)-1 receptor cytoplasmic domain of TLR4 leading to activation of TLR-mediated signalling. LPS stimulates production of reactive oxygen and nitrogen species (ROS and RNS, respectively) by activating nicotinamide adenine dinucleotide phosphate oxidase, nitric oxide-synthase, xanthine oxidoreductase and other enzymes, which leads to cellular oxidative stress. As a result, activation of redox-sensitive signalling cascades, including nuclear factor (NF)-κB, phosphorylated (p) extracellular signal regulated kinase (Erk)1/2 and p-p38 mitogen-activated protein kinases (MAPKs), triggers cellular responses to LPS, such as elevated expression of pro-inflammatory cytokines, cytoskeletal remodelling and disruption of endothelial monolayer integrity leading to acute lung injury. Amifostine reduces LPS-induced inflammatory signalling via inactivation of ROS and RNS and blunting the redox-sensitive inflammatory signalling. Iκβ: inhibitor of NF-κB; Iκκ: inhibitor of Iκβ; MEK: mitogen-activated protein kinase kinase; MAPKAPK: MAPK-activated protein kinase; Hsp27: heat shock protein 27; MLC: myosin light chain.
Several lines of evidence suggest that the endothelium is a major site of amifostine absorbance and conversion to its active form. WR-2721 is presumably modified by membrane-bound alkaline phosphatase, which is highly expressed in the endothelium and transferred into the thiol metabolite WR-1065. WR-1065 quickly penetrates into the cell, where the thiol groups act as free-radical scavengers and protect cells from oxidative damage 26. However, pulmonary endothelium and epithelium are the major sources of oxidants in the lung and possibly significant contributors in maintaining the oxidant-rich environment at the inflammatory locus. Based on the current data, the present authors speculate that the potent protective effects of amifostine in the LPS-induced vascular leak may be attributed to its local activation by lung microvascular endothelium in the inflamed lungs, leading to an increased capacity to prevent lung microvascular endothelium and alveolar epithelium from attack by oxidants.
Amifostine is a negative charged thiol, as compared to the neutral charge of NAC, which allows amifostine accumulation within the mitochondria and around the negatively charged DNA. This effect is called the net charge of thiols model. Negatively charged thiols possess higher protective potential than neutral or positively charged thiols 27. Indeed, amifostine exhibited direct and more potent effects in cells during radiation exposure than NAC cytoprotective 28. The second protective mechanism of amifostine and NAC action is the ability to activate NF-κB by virtue of their thiol group and enhance gene expression of the intracellular antioxidant enzyme manganese superoxide dismutlase (MnSOD). Amifostine exhibits higher efficiency in stimulating MnSOD expression than NAC on an equal molar basis 5. As a result, this activation leads to enhanced resistance for radiation-induced ROS and may also contribute to the reduction of oxidative stress in chronic inflammatory lung injury. These findings strongly suggest that amifostine may exhibit more potent protective effects than NAC in the clinical settings associated with pathological ROS production accompanying LPS-induced lung injury. In contrast to NAC, amifostine has been approved by the FDA for radioprotective and cytoprotective treatment in the course of radiation therapy treatments. These features make amifostine a more attractive compound for potential clinical testing.
In summary, the present study demonstrates the potent protective effects of amifostine pre-treatment in the models of LPS-induced lung injury in vivo and pulmonary EC barrier dysfunction in vitro via attenuation of oxidative stress, inhibition of redox-sensitive MAP kinases, the NF-κB inflammatory cascade, and attenuation of LPS-induced cytoskeletal remodelling and disruption of EC adhesions leading to the preservation of EC monolayer integrity (fig. 8⇑). Amifostine is currently used as a cytoprotective compound against the toxic effects of ionising radiation 7, 27.
Although rigorous testing of various protocols of amifostine administration is still required, the results of the present study suggest, for the first time, that amifostine may be considered as a preventive treatment for a wider spectrum of acute lung injury syndromes associated with the pathological elevation of reactive oxygen species production and oxidative stress, i.e. lipopolysaccharide-induced inflammation and ischaemia/reperfusion.
Support statement
This work was supported by grants from the ALA Carrier Investigator Award for K.G. Birukov; NIH/NHLBI PO1-058064 for K.G. Birukov and J.G.N. Garcia; and NIH/NCI RO1 CA99005 and DDE Grant DE-FG02-05ER64086 for D.J. Grdina.
Statement of interest
None declared.
Acknowledgments
The authors would like to thank N. Moldobaeva for superior technical assistance.
- Received January 30, 2008.
- Accepted October 8, 2008.
- © ERS Journals Ltd