



Modulation of ozone-induced airway hyperresponsiveness and inflammation by interleukin-13
- A. S. Williams 1 ,
- P. Nath 1 ,
- S-Y. Leung 1 ,
- N. Khorasani 1 ,
- A. N. J. McKenzie 2 ,
- I. M. Adcock 1 and
- K. F. Chung 1
- 1Experimental Studies, Airway Disease Section, National Heart and Lung Institute, Imperial College London, London, and 2The Medical Research Council, Laboratory of Molecular Biology, Cambridge, UK.
- K. F. Chung, Experimental Studies, Airway Disease Section, National Heart & Lung Institute, Guy Scadding Building, Dovehouse St., London SW3 6LY, UK. Fax: 44 2073518126. E-mail: f.chung{at}imperial.ac.uk
Abstract
The present study aimed to determine whether the T-helper cell type 2-derived cytokines, interleukin (IL)-4 and -13, can modulate the lung response to ozone exposure.
IL-13-/-, IL-4/13-/- and IL-13-overexpressing transgenic (Tg) mice were exposed to ozone (3 ppm; 3 h) or air. Wild-type (Wt) Balb/c mice and transgenic-negative littermates (IL-13Wt) were used as controls for gene-deficient and IL-13Tg mice, respectively.
IL-4/13-/- and IL-13-/- mice developed a lesser degree of ozone-induced airway hyperresponsiveness (AHR) while IL-13Tg mice developed a greater degree of AHR compared with ozone-exposed wild-type or IL-13Wt mice, respectively. Ozone caused a time-dependent increase of bronchoalveolar lavage (BAL) neutrophils and macrophages in wild-type mice, maximal at 20–24 h, which was attenuated in the IL-13-/- and IL-4/13-/- mice. In IL-13Tg mice, there was a greater increase in BAL neutrophils after ozone exposure compared with IL-13Wt mice. Using quantitative real-time PCR, ozone-induced mRNA expression for IL-6 and keratinocyte chemokine was further enhanced in IL-13-/- and IL-4/13-/- mice, and was inhibited in IL-13Tg mice. Macrophage inflammatory protein (MIP)-3α/CCL20 expression was enhanced after ozone exposure in wild-type mice, inhibited in IL-13-/- and IL-4/13-/- mice, while in IL-13Tg mice it was enhanced. A similar pattern of expression was observed with lipopolysaccharide-induced cytokine (LIX/CXCL5/ENA-78) expression.
In conclusion, interleukin-13 augments ozone-induced airway hyperresponsiveness and neutrophilic inflammation, possibly through modulation of certain cytokines induced by ozone exposure.
- Airways hyperresponsiveness
- interleukin-4
- interleukin-13
- neutrophils
- ozone
High levels of ambient ozone, a powerful oxidant pollutant gas, pose a significant threat to respiratory health and are linked to the worsening of symptoms and increased hospitalisations of patients with obstructive lung disease, such as asthma and chronic obstructive pulmonary disease 1–7. Ozone exposure induces airway hyperresponsiveness (AHR) to bronchoconstrictor agents and lung neutrophilia in many species, including humans 8–13. The mechanisms underlying ozone-induced increase in lung resistance (RL) and inflammation are unclear. The influx of neutrophils may be due to their recruitment by pro-inflammatory cytokines and chemokines known to be upregulated by exposure to ozone in both rodents or humans, such as cytokine-induced neutrophil chemoattractant, macrophage inflammatory protein (MIP)-2 (CXCL2), tumour necrosis factor α, interleukin (IL)-1β, IL-8 (CXCL8), keratinocyte chemokine (KC; CXCL1) and IL-6 14–19. In addition, there is increased expression of MIP-3αCCL20 and lipopolysaccharide-induced CXC chemokine (LIX; CXCL5) in lungs of mice exposed to ozone (by gene microarray analysis; data not shown).
Several studies have indicated that ozone exposure may induce or amplify the allergic response. Thus, ozone exposure of healthy or allergic asthmatic individuals can lead to an eosinophilic inflammation in the upper airways 20. Furthermore, ozone exposure can increase the response of the lower airways of asthma patients to the airway or eosinophilic response to allergen 21, 22. In animal models, positive interactions between ozone exposure and components of the allergic response have also been reported 23. In the current study, the interaction of ozone with the asthmatic response was investigated by examining the role of IL-4 and -13, two structurally related cytokines involved in T-helper cell type 2-mediated immune responses 24, in the lung response to ozone exposure. IL-13 has been particularly implicated in causing many aspects of asthma including AHR 25, airway inflammation 26 and airway wall remodelling 27. The present approach was to study the effect of ozone exposure in mice in which either IL-13 alone or both IL-4 and -13 have been disrupted, and in mice in which IL-13 has been overexpressed. The present data indicate that IL-13 is an important modulator of ozone-induced AHR and lung inflammation.
METHODS
Generation of IL-13-/-, double IL-4/13-/- and IL-13Tg mice
IL-13-/- mice were created by the disruption of exon 1 in the mouse IL-13 gene 28. IL-4/13-/- mice were generated by the simultaneous disruption of the mouse IL-13 and IL-4 genes, as reported previously 24. Knockout mice were identified using PCR analysis of genomic DNA, and wild-type (Wt) Balb/c mice were used as controls. Transgenic (Tg) mice over-expressing IL-13 (IL-13Tg) were created by cloning a 6 kb BamHI-digested DNA fragment of the complete IL-13 gene upstream of the human cluster of differentiation 2 locus control region 29. Transgenic mice were identified using PCR analysis of genomic DNA and negative-transgenic littermates (IL-13Wt) were used as controls.
Study design and methods
Two separate studies were performed. In the first study, IL-13-/- and IL-4/13-/- and wild-type Balb/c mice (Harlan, Bicester, Oxon, UK) were investigated; in the second study, IL-13Tg and IL-13Wt were examined. In both studies, groups of mice were exposed to either ozone (3 ppm, 3 h) or air. Mice were exposed to ozone produced by an ozonizer (Model 500 Ozoniser; Sander, Wuppertal, Germany), mixed with air for 3 h at a concentration of 3 ppm within a sealed Perspex® container. Ozone concentration was continually monitored with an ozone probe (ATi Technologies, Oldham, UK). Bronchoalveolar lavage (BAL) was performed 3 h after exposure and lung tissues were collected for mRNA extraction for real-time PCR. Other groups of mice were studied at 20–24 h after exposure with measurement of RL and airway responsiveness to acetylcholine (ACh; Sigma, Dorset, UK), followed by BAL.
Assessment of AHR
At 20–24 h after exposure, mice were anesthetised with an intraperitoneal injection of anesthetic solution containing midazolam and hypnorm (0.315 mg·mL−1 fentanyl citrate and 10 mg·mL−1 fluanisone). Mice were tracheostomised and ventilated (250 breaths·min−1 with tidal volume of 250 μL). Mice were monitored in a whole body plethysmograph with a pneumotachograph connected to a transducer (EMMS, Bordon, UK). Transpulmonary pressure was assessed using an oesophageal catheter (EMMS). Instantaneous calculation of RL was obtained. Increasing concentrations (4–256 mg·mL−1) of ACh were administered with an ultrasonic nebuliser, and RL was recorded for a 5-min period following each concentration. RL after each concentration was expressed as percentage change from baseline RL measured following nebulised PBS. The provocative concentration of ACh required to increase RL by 150% from baseline (PC150) was calculated.
BAL
Briefly, either 3 or 20–24 h after exposure, following an overdose of anesthetic, mice were lavaged with six 0.5-mL aliquots of PBS via the endotracheal tube, which was retrieved as the BAL fluid 30. Total cell counts and differential cell counts from cytospin preparations stained by May-Grünwald-Giemsa stain were determined under an optical microscope (Olympus BH2; Olympus Optical Company Ltd, Tokyo, Japan). At least 400 cells were counted and identified as macrophages, eosinophils, lymphocytes or neutrophils according to standard morphology.
cDNA synthesis, reverse transcription and real-time PCR
Whole-lung tissue samples were homogenised under liquid nitrogen and RNA was extracted using RNeasy mini kit (Qiagen, Crawley, UK). RNA (5 µg per sample) was used to synthesise single-stranded complimentary DNA (cDNA) using random hexamers and an avian myeloblastosis virus reverse transcriptase (Promega, Southampton, UK). The cDNA generated was used as a template in subsequent real-time PCR analyses. Transcript levels were determined by real-time PCR (Rotor Gene 3000; Corbett Research, Sydney, Australia) using SYBR Green PCR Master Mix Reagent (Qiagen). The murine forward and reverse primers (0.5 µM) used are specified in table 1⇓. Cycling conditions were as follows. Step 1: 15 min at 95°C; step 2: 20 s at 94°C; step 3: 20 s at 60°C; step 4: 20 s at 72°C, with step 2 to step 4 repeated for 45 cycles. The standard curves used to determine relative expression for each primer were obtained by performing real-time PCR for a diluted sample, for example, to 1:1, 1:10, 1:100 and 1:1000. Gene expression was normalised to glyceraldehyde 3-phosphate dehydrogenase.
Real-time PCR primers
Measurement of BAL IL-6 and KC levels
Levels of IL-6 and KC were measured in BAL fluid using a DuoSet ELISA kit (R&D Systems, Abington, UK) according to the manufacturer’s instructions.
Data analysis
Data are presented as mean±sem. For multiple comparisons of different groups, the Kruskall–Wallis test for ANOVA was used, followed by the Dunn’s test for comparison between two individual groups or the Mann–Whitney test. A p-value <0.05 was considered significant.
RESULTS
AHR
There were no significant differences in the baseline RL values following PBS challenge in each experimental group. Following ozone exposure, there was an increase in airway responsiveness in all groups as denoted by a shift of the concentration response curve to the left (fig. 1a⇓ and b). However, IL-4/13-/- and IL-13-/- mice developed a lesser degree of AHR when compared with wild-type mice. In IL-13-/- mice, the increase in airway responsiveness was particularly attenuated (-log PC150 1.083±0.1 and 1.58±0.1 after ozone in wild-type and IL-13-/- mice, respectively; p<0.001; fig. 1c⇓). In all three strains, ozone induced a significant degree of AHR. Conversely, AHR after ozone exposure in IL-13Tg mice was significantly increased compared with ozone-exposed wild-type mice (-logPC150 of 0.957±0.04 and 1.239±0.09 for IL-13Tg and IL-13Wt, respectively; p<0.01; fig. 1d⇓).

Concentration–response curves in response to a, b) acetylcholine (ACh) and c, d) -log provocative concentration of ACh required to increase lung resistance (RL) by 150% from baseline (PC150) of interleukin (IL)-13-/-, IL-4/13-/-, wild-type, IL-13+/+ and IL-13Wt mice exposed to air or to ozone. Data are presented as mean + sem (a and b). ——: mean; *: p<0.05; ***; p<0.001 compared with air-exposed mice of the same strain; #: p<0.05, ##: p<0.01 compared with ozone-exposed wild-type mice.
Lung inflammation
Exposure to ozone caused a time-dependent increase in total cells, neutrophils and macrophages recovered in BAL fluid in wild-type mice, with a maximal effect at 20–24 h (figs 2⇓ and 3⇓). There was a significant time-dependent attenuation of the total numbers of BAL cells observed at 20–24 h after ozone exposure but not after 3 h in IL-13-/- and IL-4/13-/- mice (fig. 2⇓). Conversely, in IL-13+/+ transgenic mice, the total number of BAL cells was significantly higher than that observed in IL-13Wt mice at both 3 and 20–24 h after ozone exposure (fig. 2⇓). The numbers of neutrophils in the IL-13-/- mice following ozone exposure at 20–24 h were also significantly reduced compared with wild-type mice; this was also similar in IL-4/13-/- mice at 20–24 h after exposure to ozone (fig. 3b⇓). In IL-13+/+ transgenic mice, the neutrophil counts were increased at 20–24 h after ozone exposure compared with IL-13Wt mice (fig. 3b⇓). In IL-4/13-/- mice, the decrease in BAL neutrophils at 20–24 h after ozone exposure was not significantly decreased compared with wild-type mice. BAL macrophage numbers were increased at 20–24 h after ozone exposure in wild-type or IL-13Wt mice; this effect was attenuated at 24 h after ozone exposure in IL-13-/- and IL-4/13-/- mice (fig. 3a⇓). In IL-13+/+ mice, the BAL macrophage numbers were not increased compared with IL-13Wt mice (fig. 3a⇓).

Total bronchoalveolar lavage (BAL) cells after exposure to air (□: 3 h; ░: 20–24 h) or ozone (▪: 3 h; ▒: 20–24 h) in wild-type (Wt), interleukin (IL)-13-/-, IL-4/13-/-, IL-13Wt and IL-13+/+ mice. Data are presented as mean + sem. *: p<0.05; **: p<0.01; ***: p<0.001 compared with air-exposed mice of same strain at same time-point; #: p = 0.05 compared with ozone-exposed wild-type mice at same time-point; ¶: p = 0.05 compared with ozone-exposed wild-type mice at same time-point.
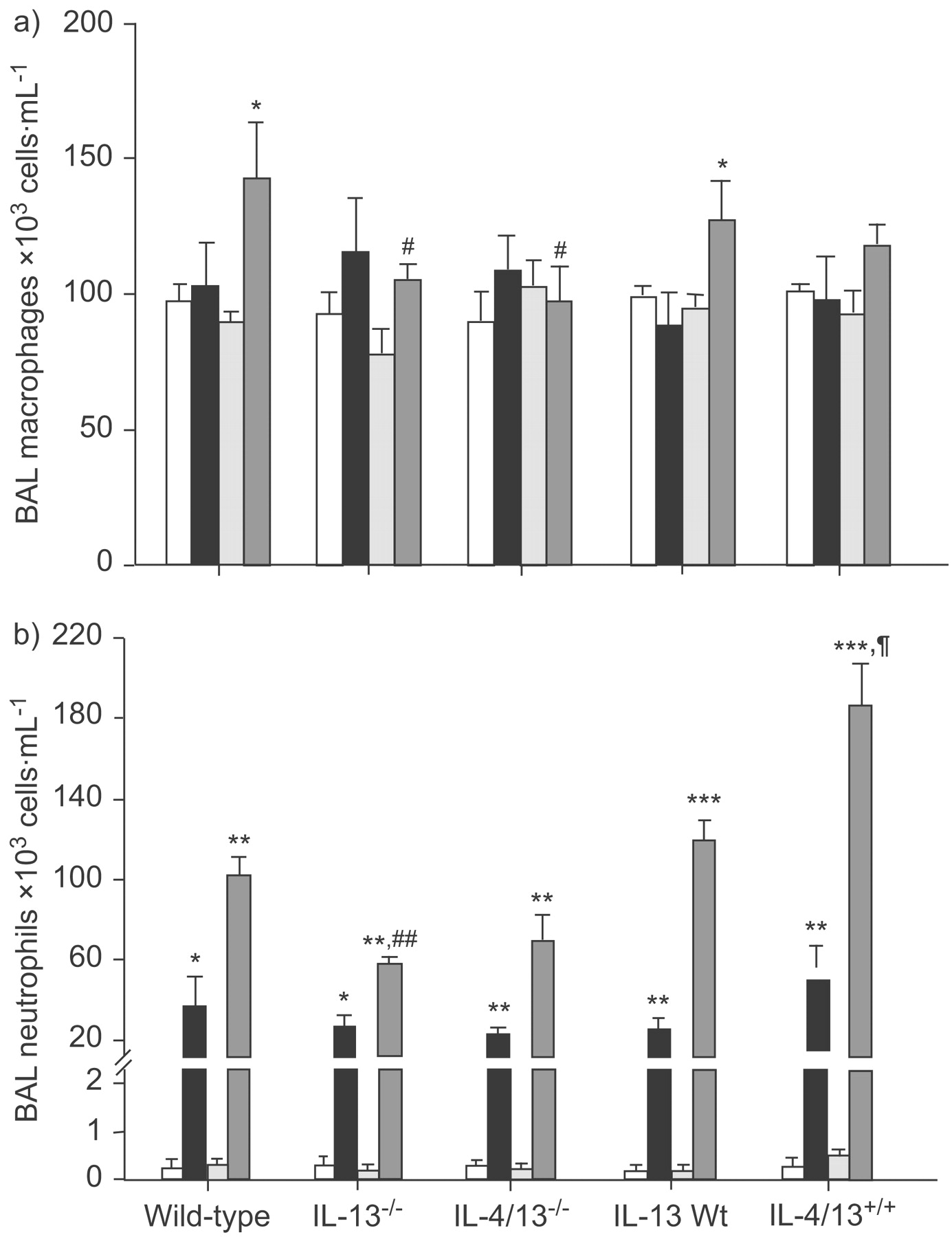
Bronchoalveolar lavage (BAL) a) macrophage and b) neutrophil cell numbers after exposure to air (□: 3 h; ░: 20–24 h) or ozone (▪: 3 h; ▒: 20–24 h) in wild-type (Wt), interleukin (IL)-13-/-, IL-4/13-/-, IL-13Wt and IL-13+/+ mice. Data are presented as mean + sem. *: p<0.05; **: p<0.01; ***: p<0.001 compared with air-exposed mice of same strain at same time-point. #: p<0.05; ##: p<0.01 compared with ozone-exposed wild-type mice at same time-point. +: p = 0.05 compared with ozone-exposed wild-type mice at same time-point.
Real-time PCR
Quantitative real-time PCR was performed in lungs from IL-13-/-, IL-4/13-/- and in IL-13Tg mice collected at 3 h after ozone exposure. There was significantly increased mRNA expression for IL-6, KC/CXCL1, MIP-3α/CCL20 and LIX/CXCL5/ENA-78 in ozone-exposed wild-type and IL-13Wt mice (fig. 4⇓). In ozone-exposed IL-13-/- and IL-4/13-/- mice, mRNA expression for IL-6 was further increased but this was not significant (fig. 4a⇓); the expression was nonsignificantly different in IL-13Tg mice, compared with respective ozone-exposed IL-13Wt controls (fig. 4a⇓). A similar pattern was observed with ozone-induced expression of KC/CXCL1. KC/CXCL1 mRNA expression was significantly less in ozone-exposed IL-13Tg mice compared with IL-13Wt mice (fig. 4d⇓); however, there was no significant increase in KC in IL-13-/- and IL-4/13-/- mice after ozone exposure (fig. 4c⇓). MIP-3α/CCL20 expression was not significantly different in IL-13-/- and IL-4/13-/- mice after ozone exposure compared with wild-type mice (fig. 4b⇓) but LIX/CXCL5/ENA-78 expression in ozone-exposed knockout mice was less than in ozone-exposed wild-type mice (p<0.05 in IL-4/13-/- and p = 0.057 in IL-4-/- mice; fig. 4e⇓). Ozone-induced MIP-3α/CCL20 and CXCL5/LIX/ENA-78 expression was nonsignificantly changed in IL-13Tg mice, compared with IL-13Wt (fig. 4b⇓ and e).
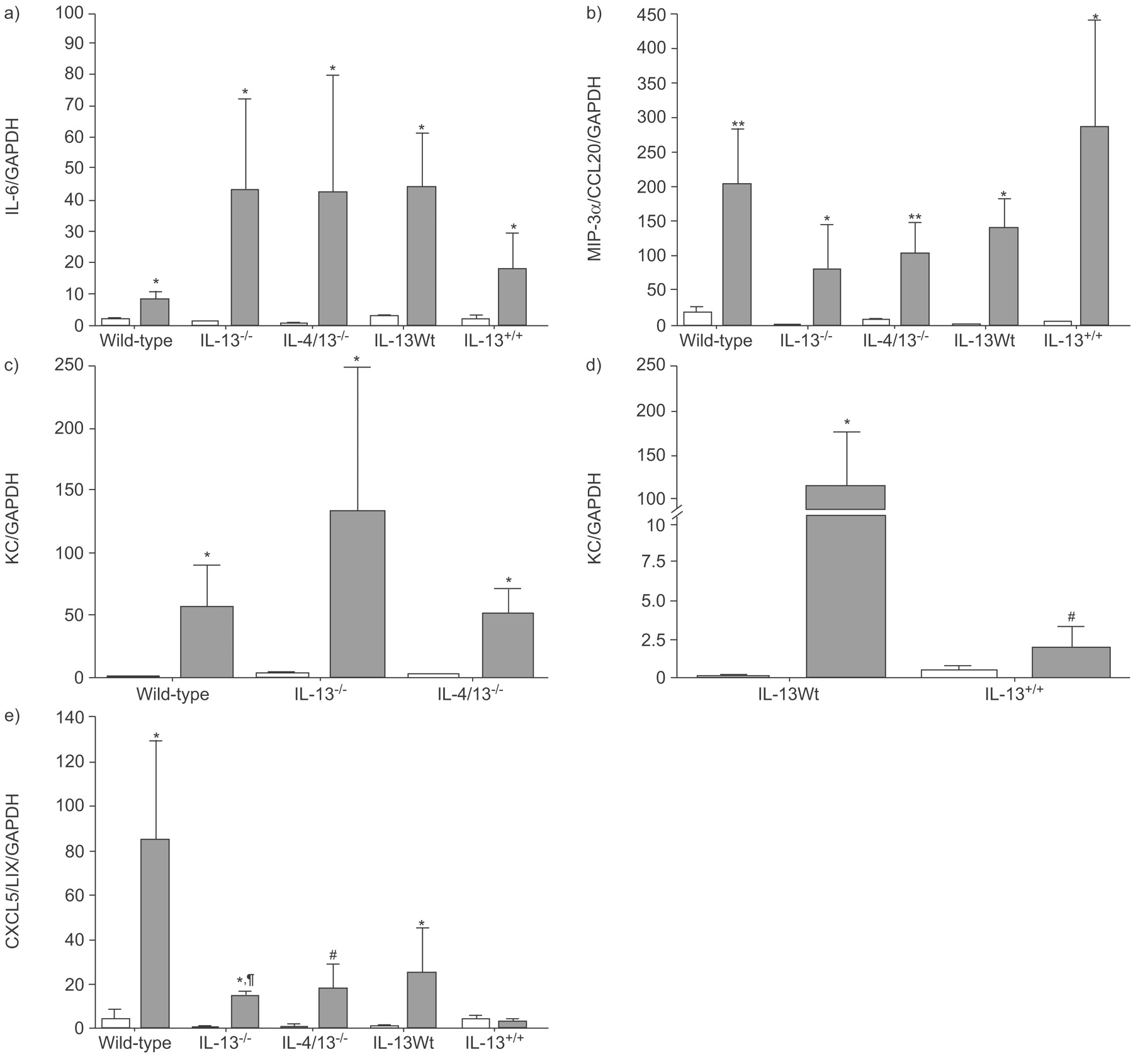
mRNA expression of a) interleukin (IL)-6, b) Macrophage inflammatory protein (MIP)-3α/CCL20 c,d) keratinocyte chemokine (KC), and e) lipopolysaccharide-induced CXC chemokine (CXCL5/LIX) expressed as a ratio of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), after exposure to air (□) or ozone (▓:) in wild-type (Wt), IL-13-/-, IL-4/13-/-, IL-13Wt and IL-13+/+ mice. Data are presented as mean + sem. *: p<0.05; **: p<0.01 compared with air-exposed wild-type mice; #: p<0.05; ¶: p = 0.057 compared with ozone-exposed wild-type mice.
BAL ELISA for IL-6 and KC
BAL levels of IL-6 and KC increased significantly after ozone exposure at 24 h (fig. 5a⇓ and b). In both IL-13-/- and IL-4/13-/- mice, the levels of IL-6 and KC were increased compared with those in wild-type mice, but the difference was only significant for IL-6 levels in IL-13-/- mice (fig. 5a⇓ and b). In IL-13+/+ transgenic mice, the level of IL-6 was not significantly lower after ozone compared with IL-13Wt mice (fig. 5a⇓); the level of KC was lower but this was not significant (fig. 5b⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bronchoalveolar (BAL) levels of a) interleukin (IL)-6 and b) keratinocyte chemokine (KC) after exposure to air (□) or ozone (▓:) in wild-type (Wt), interleukin (IL)-13-/-, IL-4/13-/-, IL-13Wt and IL-13+/+ mice. Data are presented as mean + sem. *: p<0.05; **: p<0.01 compared with air-exposed wild-type mice; #: p<0.05 compared with ozone-exposed wild-type mice.
DISCUSSION
In the present study, deletion of IL-13 or of IL-4 and IL-13 in mice was shown to protect against ozone-induced AHR and inhibit neutrophil and macrophage accumulation in the BAL fluid. By contrast, IL-13 overexpression exacerbated ozone-induced AHR and lung neutrophilia. Additionally, it was shown that ozone-induced expression of IL-6, KC/CXCL1, CCL20/MIP-3α and CXCL5/LIX were influenced by deletion of IL-13 or by overexpression of IL-13. Therefore, IL-13 can modulate the airway and inflammatory response to ozone. The contribution of IL-4 was unclear from the results observed in IL-4/13-/- mice, and can only be resolved by studying IL-4-/- mice.
The mechanisms of ozone-induced AHR are unknown but it is possible that ozone may directly cause an increase in airway smooth muscle (ASM) contractility to form the basis of AHR. Preliminary data from the present authors’ laboratory has shown that bronchi from ozone-exposed mice show increased isometric-contractile responses to acetylcholine ex vivo and that this increased contractility together with the increased AHR are abolished by pre-treatment of mice to an antioxidant, N-acetylcysteine (data not shown; F.X. Blanc, Imperial College, London, UK; personal communication). Similarly, the effect of IL-13 in increasing AHR may be through a direct action on the ASM, since IL-13 has been shown to directly increase ASM contractility through the activation of IL-13 receptors identified on ASM cells 31. IL-13 increases carbachol- and KCl-induced maximal force of murine tracheal rings 32. IL-13 also reduces isoproterenol-induced relaxation as measured by cell stiffness in human ASM cells 31. The potential for IL-13 to increase bronchial responsiveness in vivo has also been demonstrated in mice where direct instillation of IL-13 into the lungs causes AHR 33. In addition, in IL-13-/- mice, allergen-induced AHR is inhibited 34, while in IL-13Tg mice, it is enhanced 25.
Interestingly, the present observations support the notion that IL-13 positively influenced the degree of ozone-induced lung neutrophilia. IL-13 is not a known chemoattractant for neutrophils but for eosinophils; this is an effect that may be mediated through the release of the eosinophil chemokine, eotaxin/CCL13 35, 36. It has been reported that patients with allergic rhinitis exposed to ozone experimentally produce an eosinophilic response 37, 38. It is possible that the lack of an eosinophilic response in the mice is due to the fact that the bone marrow is not primed to produce eosinophils, particularly in the IL-13-/- mice. One mechanism for the increased neutrophilic response may relate to the potential for IL-13 to increase oxidative stress in the lungs, thus contributing to the oxidant stimulus of ozone. IL-13 and IL-4 are potent inducers of nicotinamide adenine dinucleotide phosphate oxidase enzyme, dual oxidase-1, in the airway epithelium 39. IL-13 enhances blood monocyte superoxide production in response to phorbol myristate acetate 40; however, another study has indicated that IL-13 inhibits protein kinase C-triggered respiratory burst in human monocytes 41. More recently, IL-13 has been reported to increase superoxide production from human bronchial epithelial cells 42. Therefore, the possibility that IL-13 can contribute to oxidative stress or worsen the consequences of oxidative stress deserves further study in the present model. There may also be other mechanisms of enhancement including at the level of tissue permeability or T-cell activation.
The present data regarding the measurement of cytokine expression in the lungs of ozone-exposed mice may provide a clue as to the mechanisms by which IL-13 may modulate ozone-induced AHR and inflammation. These results show a complex pattern of changes of the level of expression of these cytokines, but must be interpreted as preliminary since they were examined at only one time-point (3 h after exposure) and there was variability in the responses that precluded significance in many of the comparisons using numbers of observations that were sufficiently powerful to answer the primary questions on AHR and inflammation. In the present study, only four of many cytokines that are known to be upregulated by ozone exposure were chosen, namely the pro-inflammatory cytokines KC and LIX/CXCL5/ENA-78, both neutrophil chemoattractants and activators, while IL-6 has pro- and anti-inflammatory activities and MIP-3α/CCL20 is a chemoattractant for dendritic cells.
Using quantitative real-time PCR, the present authors showed that there was a nonsignificant increase in IL-6 and KC/CXCL1 expression in IL-13-/- mice, while LIX/CXCL5/ENA-78 was significantly inhibited in IL-13-/- and IL-4/13-/- mice after ozone-exposure. IL-6 expression was decreased nonsignificantly in IL-13+/+ mice after ozone exposure. The modulation of IL-6 gene expression, observed in the knock-out and transgenic strains, was similar to that observed of IL-6 protein levels in BAL fluid. These results suggest that IL-13 expression may modulate ozone-induced expression of IL-6. Previous studies have shown that bradykinin-induced increase in IL-6 protein and mRNA is inhibited by IL-13 in human ASM cells and that IL-6 expression was also found to be bradykinin B2 receptor-dependent 43. Interestingly, IL-6 has been shown to play a pro-inflammatory role in ozone-induced inflammation 44.
In conclusion, the present authors demonstrate an important role for interleukin-13 in ozone-induced airway hyperresponsiveness and inflammation. Inhibition of interleukin-13 may, therefore, prevent the inflammation and airway hyperresponsiveness in nonallergic airway diseases where oxidative stress is a key component, such as in asthma and chronic obstructive pulmonary disease.
Support statement
This study was funded by a grant from Imperial College Trust. K.F. Chung was supported by a Wellcome Trust programme grant.
Statement of interest
None declared.
- Received September 14, 2007.
- Accepted March 28, 2008.
- © ERS Journals Ltd