



Levofloxacin versus clarithromycin in COPD exacerbation: focus on exacerbation-free interval
- H. Lode,
- J. Eller,
- A. Linnhoff,
- M. Ioanas and
- the Evaluation of Therapy-Free Interval in COPD Patients Study Group
- H. Lode, Helios Klinikum Emil von Behring, Zum Heckeshorn 33, D-14109 Berlin, Germany. Fax: 49 3080022623. E-mail: haloheck@zedat.fu-berlin.de
Abstract
Antibiotic treatment of bacterial exacerbation of chronic obstructive pulmonary disease (COPD) shows some immediate clinical benefits and may also minimise the frequency of further recurrences.
Patients (n=511) were enrolled into a randomised double-blind multicentric study comparing the exacerbation-free interval (EFI), efficacy and safety of 7-day levofloxacin versus 10-day clarithromycin in patients with COPD exacerbation. Patients were monitored over a 1-yr period. A total of 434 patients (per protocol population) received the medication for ≥5 days.
The median EFI in the per protocol population was 300 days for levofloxacin and 350 days for clarithromycin. For patients with a new documented exacerbation during follow-up (n=223), the median EFI was 100.5 days in the levofloxacin group and 95 days for clarithromycin. No significant differences in EFI between groups could be observed when stratifying the study population according to microbial aetiology and severity of bronchial obstruction. Levofloxacin and clarithromycin showed similar clinical success rates. The bacteriological success rate was significantly higher in the levofloxacin group. Both antibiotics were well tolerated.
In summary, levofloxacin was associated with a significantly higher bacteriological eradication rate but similar exacerbation-free interval in patients with chronic obstructive pulmonary disease exacerbation compared to clarithromycin.
- Chronic obstructive pulmonary disease
- clarithromycin
- exacerbation
- exacerbation-free interval
- levofloxacin
This study was supported by a grant from Aventis Pharma, Bad Soden, Germany.
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are typical events that characterise the course of the disease and are the most common cause of death in these patients 1. Exacerbations result in impaired quality of life, decline in lung function and increased health care utilisation 3–5. Although the aetiology of COPD exacerbation has not been completely clarified, there is strong evidence that potentially pathogenic microorganisms (PPMs) are isolated from at least half of COPD patients during exacerbations 6–9. Another 30–40% of the exacerbations of COPD were recently shown to be attributable to viruses 10.
In this context, antimicrobial therapy remains a controversial issue, although it shows some immediate clinical benefits compared to no therapy 12. A clear indication for antibiotic treatment appears to be sputum purulence, a simple parameter for discriminating between bacterial and nonbacterial exacerbation 13.
Fluoroquinolones seem to be an adequate choice, taking into account their bactericidal activity in vitro against most of the pathogens involved in COPD exacerbation, including penicillin-resistant Streptococcus pneumoniae (gatifloxacin, moxifloxacin, levofloxacin and gemifloxacin) and Pseudomonas aeruginosa (ciprofloxacin). Furthermore, the good penetration into lung tissue and respiratory secretions, one-dosage daily administration (for the new quinolones) and short duration of treatment also favour choice of these drugs in COPD exacerbation.
Several clinical trials support the clinical efficacy of fluoroquinolones in exacerbation of chronic bronchitis 14 compared to macrolides or β-lactams. Moreover, the recent study of Wilson et al. 16 shows that fluoroquinolones (i.e. gemifloxacin) exhibit an additional benefit compared to macrolides (i.e. clarithromycin), in that they reduce the recurrence of exacerbations. Since fluoroquinolones and macrolides seem to exhibit rather comparable clinical and bacteriological efficacy, as well as similar safety profiles 14, this finding may have considerable impact on therapeutic choice, especially in COPD patients with frequent exacerbations.
Based on these data, the aim of the present study was to compare the exacerbation-free interval (EFI) following treatment with levofloxacin and clarithromycin in COPD exacerbation. Several clinical trials have demonstrated that levofloxacin shows clinical and bacteriological efficacy inacute exacerbation of chronic bronchitis 14. Clarithromycin was used as comparator because of its proven efficacy in this condition 16. Secondary objectives included comparisons of clinical and bacteriological response, as well as the safety profile of the two antibiotics.
Methods
Study design and patients
The current prospective randomised multicentric double-blind comparative study was performed using a double-dummy design with two-arm parallel groups. Patients were randomised in a 1:1 ratio to receive either oral levofloxacin (Aventis Pharma Deutschland, Bad Soden, Germany) 500 mg once daily for 7 days followed by placebo (Allphamed PHARBIL Arzneimittel, Göttingen, Germany) for 3 days, or clarithromycin (Abbott, Wiesbaden, Germany) 250 mg twice daily (the officially approved dosage of clarithromycin for this condition in Germany) for 10 days.
The study included outpatients aged >35 yrs with a clinical history of chronic bronchitis with ≥2 exacerbations·yr−1 20, a forced expiratory volume in one second (FEV1) in the range 35–75% predicted 21 and presenting an acute exacerbation. The last available FEV1 measurement in the stable state within the previous 6 months was considered for the inclusion criteria. The exacerbation was defined according to Winnipeg criteria (increased dyspnoea, increased sputum volume and purulent sputum) 22, and only patients meeting Winnipeg I (all three criteria) or II (two criteria present) were enrolled.
Patients were excluded from the study if they exhibited any of the following characteristics: severe respiratory infection requiring parenteral antibiotic treatment; evidence of a new pulmonary infiltrate on chest radiography; administration of another antibiotic within 72 h prior to enrolment; documented evidence of severe bronchiectasis; need for concomitant antimicrobial medication; current treatment with systemic steroids at a dose of >20 mg·day−1 prednisone or equivalent for >4 weeks; AIDS or another immunosuppressive condition; a history of epilepsy or lowered seizure threshold; previous enrolment in another study within the previous 4 weeks; pregnancy or nursing; allergy to fluoroquinolones ormacrolide derivatives; severe underlying diseases, such as cystic fibrosis, active tuberculosis, or suspected lung or chest malignancy; progressively fatal disease; malabsorption syndrome; hepatic disease; renal impairment (creatinine level >2.0 mg·dL−1); or alcohol or drug abuse.
All patients provided written informed consent and the study protocol was approved for all centres by the local ethics committees. The study was conducted according to the Good Clinical Practice Guidelines of the European Union and the Declaration of Helsinki.
Follow-up and exacerbation-free interval assessment
Patients were monitored over a period of 1 yr, with scheduled visits at weeks 6, 18, 36 and 52. When patients could not attend a scheduled visit, they were contacted by telephone. Patients were instructed to contact the investigator(s) responsible for the study immediately if there was any change in their health status. Diagnosis of a new exacerbation was based on the same clinical criteria as the previous. During each visit, patients were asked to report any event that required antibiotic therapy and/or hospitalisation elsewhere, in order to avoid unrecorded exacerbations.
In agreement with the studies of Chodosh and coworkers 15, all clinical failures during the study therapy were counted as zero EFI days. For patients with no new exacerbation during the 1-yr observation period, the EFI was considered to be the number of days that had elapsed between the index exacerbation and the time point of the last information available (censored data). In all other cases, the number of days that had elapsed between the onset of exacerbations was taken into account. For calculation, the onset of an exacerbation was considered the day of medical attendance.
Clinical and bacteriological assessments and definitions
Clinical signs and symptoms of acute exacerbation (cough, dyspnoea, sputum characteristics and auscultatory assessment) were noted before initiating the treatment, after 3–5 days and at day 11–14 (test of cure).
Any further exacerbation occurring during the follow-up period was evaluated based on the same criteria as the index episode. The clinical response at the end of treatment, with respect to the responder analysis, was defined as follows: success, if all infection-related symptoms disappeared or were improved to such an extent that no further antibiotic therapy was indicated; failure, if there was no improvement or deterioration of the symptoms; and unknown, if no assessment was possible.
Microbiological evaluation of the sputum samples was performed before treatment, at day 3–5 and, if possible, at the end of treatment (day 11–14). According to the criteria of the American Society for Microbiology 24, only sputa with <10 or 10–25 squamous oropharyngeal epithelial cells and >25 leukocytes per low power field (x100) were considered for culture. Culture was performed according to standard microbiological methods 25. Susceptibility was determined by a standard disc diffusion technique recommended by the National Committee for Clinical Laboratory Standards 26. A proven bacterial aetiology was not mandatory for study enrolment.
A satisfactory bacteriological response was defined as eradication (the baseline bacteriological pathogen was eradicated) or presumed eradication (the patient had improved clinically to such an extent that a satisfactory follow-up culture from sputum samples could not be obtained). An unsatisfactory response was recorded as persistence (the baseline causative pathogen was still present irrespective of the presence or absence of signs of infection), relapse (the absence of the baseline causative pathogen was documented but the same pathogen appeared in cultures of specimens obtained after the end of treatment) or superinfection (a new causative pathogen isolated from any site during therapy or within 3 days after treatment completion, together with clinical evidence of infection).
Safety assessment
Adverse events were evaluated in all patients that received at least one dose of the study drug (safety population). Adverse events were recorded at all visits and ranked by intensity (mild, moderate, severe and serious) and relationship to the study medication.
Statistical analysis
The sample size calculation was based on survival analysis models with a two-sided type I error probability (α) of 0.05 and a type II error probability (β) of 0.20, i.e. a power of 80%. Expecting a mean duration of 4–6 months to the next exacerbation under clarithromycin and a 50% increase in the EFI under levofloxacin and assuming, furthermore, a mean observation period of ∼6.5 months per patient, a required sample size of 251 patients per treatment group was estimated.
Survival analysis techniques were employed to calculate EFIs and the Kaplan–Meier method was used to estimate the distribution of time to failure. The Wilcoxon test and log-rank test were applied to compare the survival curves for each study drug group. The latter, which places more weight on later times of failure, was used for the formal testing of the study hypothesis (superiority of levofloxacin over clarithromycin). The median and, respectively, the 95% confidence interval (CI) of the median EFI could not be calculated for some subgroups, since <50% of the study population developed a new exacerbation over the study period; therefore, the 25th percentile has been also estimated.
The response (clinical and bacteriological) data were analysed by centre-adjusted Cochran–Mantel–Haenszeltests. In addition, two-sided 95% CIs for the treatment differences in response rates were calculated.
Comparability of baseline characteristics was descriptively analysed using the Cochran–Mantel–Haenszel test stratified by centre (categorical parameters) or analysis of variance models with the factors treatment group and centre and theinteraction term of treatment by centre (continuous parameters).
Statistical analysis was performed separately on the following populations: intention-to-treat (ITT) population, including all randomised patients; safety population, including all patients who received at least one dose of the study medication; modified-ITT (m-ITT) population, consisting of patients who received the study medication for ≥2 days; per protocol (PP) population, consisting of patients who adhered to the study protocol and received the study medication for ≥5 days; and PP microbiology population, including all PP population patients with at least one causative pathogen isolated at enrolment and a bacteriological evaluation at the end of treatment.
Results
Baseline characteristics
The study was conducted in 36 centres in Germany, and 511 patients with a diagnosis of acute exacerbation of COPD were enrolled. The distribution of the study population is indicated in figure 1⇓. As one patient refused to participate before starting treatment, a total of 510 patients were evaluable in the safety analysis (safety population). Six patients were treated for <2 days and were evaluated only in the safety population. A total of 504 patients (55% male; mean age 59.7±11.1 yrs) with a mean FEV1 of 58.1±12.1% pred measured in the stable state were included in the m-ITT analysis. The baseline characteristics of the two treatment groups were similar (table 1⇓). A total of 477 (93.5%) patients received the complete 10 days' medication (94.1% in levofloxacin group and 93.0% in clarithromycin group). The most frequent comorbid conditions in the two treatment groups were cardiovascular diseases (35.6% of patients in levofloxacin group and 38.6% in clarithromycin group) and metabolic disorders (18.8% in levofloxacin group and 21.7% in clarithromycin group). Nearly all patients, 250 in each group, received concomitant medication over the study period, consisting of inhaled corticosteroids (10.3% in levofloxacin group and 9.8% in clarithromycin group), and selective β2-agonists (20.4% in levofloxacin group and 19.8% in clarithromycin group) or xanthin-including derivatives (10.9% in levofloxacin group and 10.7% in clarithromycin group).
Drug efficacy could be evaluated in 434 patients (PP population; n=223 in levofloxacin group and n=211 inclarithromycin group). Protocol violations such as administration of drug medication for <5 days (n=24), violation of study timing (n=21) or missing data (n=12) were the primary reasons for the exclusion of 77 patients from the analysis of the PP population. Baseline characteristics and clinical respiratory signs and symptoms were similar in the PP population and the m-ITT group (data not shown).
Exacerbation-free interval
The EFIs are presented in table 2⇓. No significant differences in EFI could be observed between the two study drugs in the m-ITT and PP populations. The EFI was similar in the subgroup of patients with a new documented exacerbation and in that with a documented microbial infection at enrolment.
A similar trend in the EFI was observed in the two study groups when patients were stratified according to the presence of S. pneumoniae and Haemophilus influenzae at enrolment.
A total of 43.6% of patients in the levofloxacin group and 47.9% of patients in the clarithromycin group in the m-ITT population exhibited no relapse during the 12-month period after therapy (p=0.967).
When stratifying the patients according to the severity of obstruction (FEV1 ≥ or <50% pred), no significant differences could be observed in EFI between the two study groups (table 2⇓) or between severity groups (data not shown).
Figure 2⇓ shows the Kaplan–Meier survival analysis, revealing the EFIs for the m-ITT population with an FEV1 of <50% pred.
Clinical response
In the evaluable m-ITT population (n=433), clinical success at the end of treatment was observed in 82.8% of patients inthe levofloxacin group and 79.8% in the clarithromycin group (3.0% difference; 95% CI −4.4–10.3; p=0.428) (fig. 3⇓). Regarding the evaluable PP population (n=365), clinical success of 86.1 and 84.8% was documented inthe levofloxacin and clarithromycin groups, respectively (1.3% difference; 95% CI −6.0–8.5; p=0.732). In ∼15% of the m-ITT population and nearly 15% of the PP group, no clinical assessment was available at the end of therapy because of missing data.
Bacteriological response
PPMs responsible for the acute exacerbation were isolated in 125 (50%) patients in the levofloxacin group and 131 (51.6%) in the clarithromycin group. The most frequently isolated strains were: H. influenzae (n=80; 38 (23.9%) in levofloxacin group and 42 (25.8%) in clarithromycin group), S. pneumoniae (n=50; 23 (14.5%) in levofloxacin group and 27 (16.6%) in clarithromycin group), Moraxella catarrhalis (n=39; 22 (13.8%) in levofloxacin group and 17 (10.4%) in clarithromycin group), and Staphylococcus aureus (n=24; nine (5.7%) in levofloxacin group and 15 (9.2%) in clarithromycin group). Of the 322 strains of PPMs isolated at baseline, 34.5% were resistant to clarithromycin and only one strain (S. pneumoniae) showed an intermediate level of resistance to levofloxacin. Of the Haemophilus spp. strains, 35% were resistant to clarithromycin, whereas none was resistant to levofloxacin.
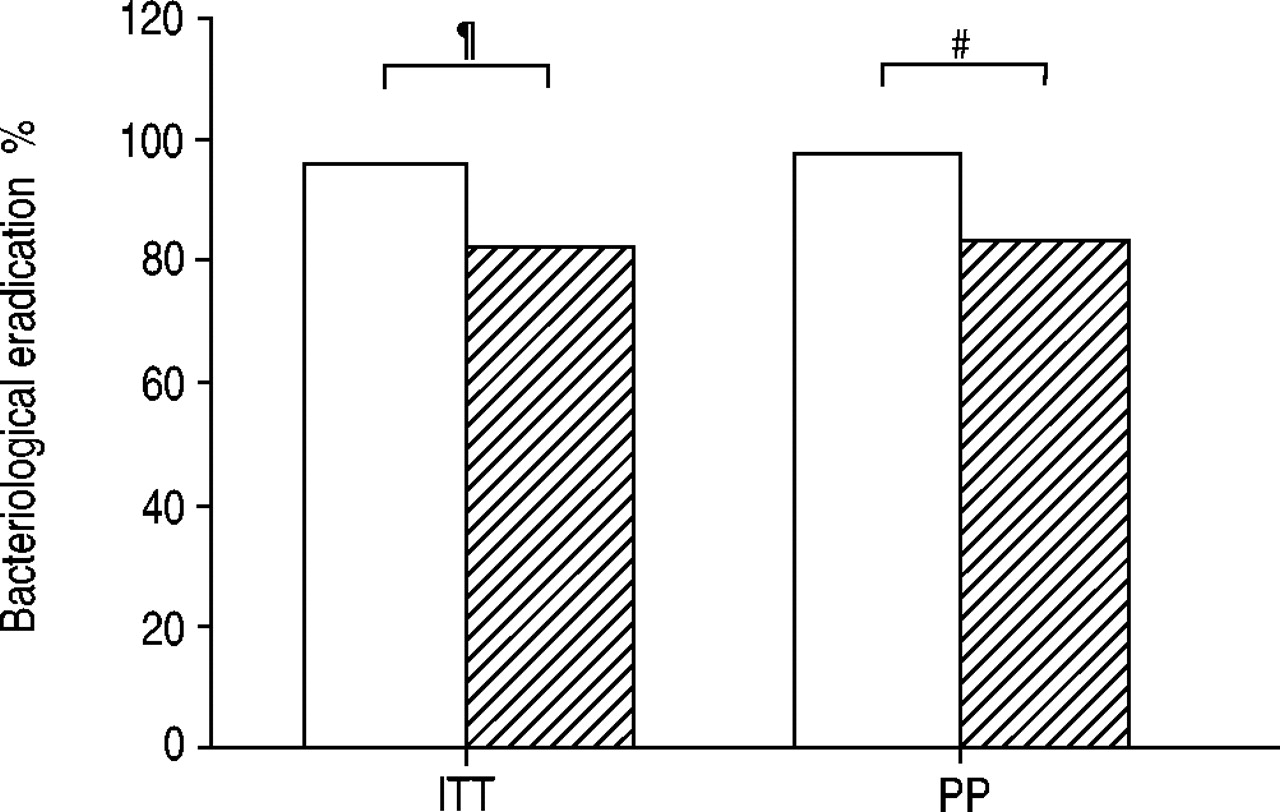
The bacteriological eradication rate at the end of treatment in the evaluable m-ITT population with microbial aetiology (n=251) was significantly higher in the levofloxacin group than in the clarithromycin group (96.0 versus 81.7%; 14.3% difference; 95% CI 6.7–21.8; p<0.0001) (fig. 4). A significantly higher bacteriological success rate in favour of levofloxacin compared to clarithromycin (96.8 versus 83.1%; 13.7% difference; 95% CI 3.7–23.8; p=0.01) was also demonstrated in the PP microbiology group (n=130).
Safety and adverse events
Forty-nine patients, 24 (9.5%) in the levofloxacin group and 25 (9.7%) in the clarithromycin group, reported 75 adverse events (31 and 44, respectively), considered to be at least possibly drug related. Most frequent were gastrointestinal adverse drug reactions (5.5% in levofloxacin group and 6.6% in clarithromycin group) and disorders of the central nervous system (0.8% in levofloxacin group and 2.3% in clarithromycin group). Most adverse events were mild to moderate. Study drug treatment was prematurely discontinued because of one or more adverse events in 26 patients (n=14 in levofloxacin group and n=12 in clarithromycin group), mostly due to gastrointestinal symptoms.
Discussion
The present study showed no difference in EFI between treatment with levofloxacin and clarithromycin in acute exacerbation of COPD. Levofloxacin was associated with a higher bacteriological success rate, but the clinical success rates were similar for levofloxacin and clarithomycin.
Acute exacerbations of COPD characterise the natural history of COPD and are related to a decline in lung function 5, deterioration of quality of life 3 and increased costs 4. Since ∼50% of all exacerbations are attributable to microbial infection 27, adequate antibiotic treatment has been shown to have beneficial effects on clinical outcome and the recurrence of acute exacerbations of chronic bronchitis 22.
The choice of empirical therapy has been facilitated by classification of the acute exacerbation and the related microbial spectrum according to the severity of bronchial obstruction, recurrence of annual exacerbations and comorbid conditions 29. Despite the constant emergence of resistance to antibiotics by H. influenzae and S. pneumoniae, recent studies report similar success rates for macrolides and fluoroquinolones 14, which are widely used in COPD exacerbation and also recommended by recent guidelines 31.
The EFI is a parameter that can make a difference when choosing antibiotic therapy, since fewer recurrences also mean a decrease in healthcare utilisation in COPD exacerbation. Destache et al. 32 reported in 1999 that patients with acute exacerbation of chronic bronchitis receiving ciprofloxacin, azithromycin or amoxicillin–clavulanate experience fewer hospitalisations and recurrences compared to those receiving older antibiotics.
A study by Wilson et al. 16 compared the long-term clinical outcome in patients with acute exacerbation of chronic bronchitis treated with gemifloxacin and clarithromycin and followed for a 26-week period. More patients receiving gemifloxacin remained free of recurrences compared to those receiving clarithromycin (71.0 versus 58.5%, p=0.016).
Another recent study comparing moxifloxacin and standard antibiotic therapy (amoxicillin, clarithromycin or cefuroxime axetil) in acute exacerbation of chronic bronchitis also found that moxifloxacin was superior in terms of clinical cure, bacteriological eradication and EFI (within 9 months of follow-up) 33. However, this study did not differentiate between the three standard comparators, and so it is difficult to conclude whether or not moxifloxacin is really superior to each of them 34.
In contrast, no significant differences were found between levofloxacin and clarithromycin regarding EFI over the 1-yr follow-up in the present study.
This discrepancy compared to the studies of Wilson and coworkers 16 could presumably be accounted for by the different follow-up periods used in the two studies: a period of<1 yr could be confounding, especially for infrequent exacerbators (<2 exacerbations·yr−1), and could result in a winter season being skipped. Another explanation may be the different nature of the exacerbation, namely whether it is due to a colonising pathogen 35 or a newly acquired microbe 6. In the first situation, the more effective bacteriological eradication demonstrated by the quinolones may genuinely be responsible for a longer stable-state period. However, the EFI is difficult to define only in terms of successful bacteriological eradication, since a considerable number of exacerbations can be caused by a new strain, as Sethi et al. 6 showed recently.
Another aspect that can make a difference in evaluating the EFI is the severity of the bronchial obstruction. A low FEV1 has been associated with more frequent exacerbations 36, and so this group is supposed to be a more adequate target for antibiotics that prolong the stable-state period. One limitation of the present study is that most (∼75%) of the patients showed a moderate degree of bronchial obstruction, which may have contributed to the low rate of exacerbations observed over 1 yr. However, similar EFIs were noted in the two severity groups irrespective of study medication.
In the present study, the bacteriological success rate was higher for levofloxacin than for clarithromycin, in relation also to the high level of in vitro resistance to clarithromycin (one-third of the H. influenzae strains were resistant to clarithromycin). Moreover, the proportion of patients with microbial growth in sputum was much higher (50%) compared to other studies 16. The differences in microbial eradication among studies are also related to the pattern of resistance of the centres involved.
However, similar clinical success rates were observed for levofloxacin and clarithromycin in the present study. One explanation for the non-inferior clinical efficacy of clarithromycin despite the considerable proportion of resistant bacteria could be the anti-inflammatory effect of macrolides in general 37, which may compensate the limited antimicrobial activity. Secondly, this contrast between invitro resistance and favourable clinical outcome may also be explained by the high penetration of macrolides in different lung compartments, including the bronchial mucosa, allowing prolonged exposure to these drugs at concentrations greater than the minimum inhibitory concentration at the infection site 38.
In conclusion, treatment with levofloxacin in acute exacerbation of chronic obstructive pulmonary disease was associated with a higher bacteriological success rate than with clarithromycin. No differences in the length of exacerbation-free interval between the two study groups were observed and the frequency of recurrences over the 1-yr follow-up was also comparable. More studies are required to clarify this aspect, because of its major implications for the course of the disease and the related costs.

Distribution of study population. ITT: intention to treat; m-ITT: modified-ITT; PP: per protocol.

Kaplan–Meier survival analysis of exacerbation-free interval in modified intention-to-treat group patients with a forced expiratory volume in one second of <50% predicted (––––: clarithromycin; ------: levofloxacin). No difference could be observed (p=0.322).

Clinical resolution at the end of therapy in the evaluable intent-to-treat (ITT) and per protocol (PP) populations of patients with chronic obstructive pulmonary disease exacerbation treated with levofloxacin (□) or clarithromycin (└).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bacteriological eradication at the end of therapy in the evaluable intent-to-treat (ITT) and per protocol (PP) populations of patients with chronic obstructive pulmonary disease exacerbation treated with levofloxacin (□) or clarithromycin (└). #: p=0.01; ¶: p<0.0001.
Baseline characteristics of the modified intention-to-treat (m-ITT) population
Exacerbation-free interval (EFI) in days
Acknowledgments
Evaluation of Therapy-Free Interval in COPD Patients Study Group members: U.W. Ballies, Kiel; E. Baumann, Munich; E. Beck, Rüdersdorf; J.C. Becker, Lübeck; A. Bisping-Arnold, Freising; H. Blaufuß, Munich; V. Brauner, Bad Soden; J. Brückner, Munich; J. Clasen, Bad Oldesloe; H.W. Fischer, Verden; R. Gebhardt, Berlin; H. Grygier, Bad Homburg; U. Harnest, Munich; H.C. Hartung, Lüdenscheid; G. Hoppe, Berlin; V. Janekovic, Frankfurt am Main; B. Kemmerich, Munich; M. Korduan, Berlin; M. Ksoll, Frankfurt am Main; J. Lehnert, Munich; H. Mauch, Berlin; D. Mernitz, Munich; H. Neppl, Celle; P. Oblinger, Munich; M. Qidan, Frankfurt am Main; H.H. Ponitz, Berlin; H. Portheine, Koblenz; B. Raack, Celle; H. Riediger, Celle; K. Roscher, Bad Soden; V. Schäfer, Frankfurt am Main; E. Scheer, Berlin; H.W. Schiware, Bremen; R. Schnorr, Berlin; H. Schriewer, Lüdenscheid; G. Semrau, Berlin; W. Stengel, Lübeck; W. Tzimas, Munich; C. Uhde, Munich; L. von Versen, Berlin; W. Weede, Munich; J. Weöres, Munich; R.W. Wepler, Ulm; and C. Witt, Berlin.
Footnotes
-
For editorial comments see page 896.
- Received January 23, 2004.
- Accepted July 16, 2004.
- © ERS Journals Ltd