



Transcription factor activation and mitogenic synergism in airway smooth muscle cells
- 1Dept of Pharmacology, University of Nebraska Medical Center, Omaha, NE, and 2Dept of Pharmacology, Emory University School of Medicine, Atlanta, GA, USA
- M.L. Toews, 986260 Nebraska Medical Center, Omaha, NE 68198‐6260, USA. Fax: 1 4025597495. E‐mail: mtoews@unmc.edu
Abstract
Simultaneous treatment of human airway smooth muscle (HASM) cells with lysophosphatidic acid (LPA) and epidermal growth factor (EGF) leads to strikingly synergistic stimulation of mitogenesis. The purpose of this study was to explore potential sites for signal integration mediating synergism, focusing on extracellular signal-regulated kinase (ERK) and transcription factors involved in proliferation and inflammation as likely candidates.
Activation of ERK was analysed by immunoblotting. Transcription factor activation was assessed using HASM cells transduced with luciferase reporter gene constructs.
LPA and EGF both activated ERK but had no synergistic effect when combined. LPA and EGF both activated activator protein (AP)‐1, cyclic adenosine monophosphate response element-binding protein, nuclear factor of activated T‐cells and the serum response element; however, only AP‐1 activation exhibited synergism. Activation of the inhibitory guanine nucleotide-binding protein and of ERK signalling pathways were required for most transcription factor responses to LPA. In contrast, nuclear factor (NF)‐κB was activated by LPA but not EGF and NF‐κB activation was completely blocked only when Rho was inhibited. Rapid activation of Rho was observed in response to LPA but not to EGF. Importantly, inhibition of Rho selectively blocked synergism in both AP‐1 activation and mitogenesis.
In summary, extracellular signal-regulated kinase activation is required for many transcription factor responses to lysophosphatidic acid and epidermal growth factor, however it is not synergistic. Activation of activator protein‐1 is synergistic, and Rho activation by lysophosphatidic acid is required for synergism in both activator protein‐1 activation and mitogenesis.
- activator protein‐1
- asthma
- growth factors
- nuclear factor‐κB
- proliferation
- Rho
This study was supported by the University of Nebraska Medical Center seed grants to M.L. Toews, a University of Nebraska Emley Fellowship to T.L. Ediger and by research grants HL52810 and HL56107 from the National Heart Lung and Blood Institute to T.J. Murphy.
Regulation of human airway smooth muscle (HASM) cell proliferation is clinically relevant to the pathology of asthma because increases in airway smooth muscle mass are seen in asthma and contribute to enhanced airway narrowing 1, 2. Increased airway smooth muscle mass is one component of airway remodelling, an important process in the pathology not only of asthma, but also of fibrotic diseases such as chronic bronchitis. Studies of highly proliferative HASM cells in culture can shed light on potential regulatory mechanisms in vivo 1.
HASM cells grown in vitro proliferate in response to a variety of stimuli, including G protein-coupled receptor (GPCR) agonists as well as peptide growth factors acting via receptor tyrosine kinases (RTKs) 1, 2. Recent studies from the present and other authors have provided evidence that lysophosphatidic acid (LPA), a phospholipid mitogen present in serum, may be an important regulator of airway cell function and have also suggested possible roles for LPA in airway disease 3. HASM cells show increased mitogenesis in response to LPA, a GPCR mitogen, and to epidermal growth factor (EGF), an RTK growth factor. Interestingly, HASM cells treated with LPA together with EGF show a strikingly synergistic stimulation of mitogenesis, much greater than the sum of the responses to the two agents alone 4, 5. Understanding the signalling pathways involved in the synergistic mitogenesis, seen when cells are treated with multiple stimuli, is an important aspect of understanding how cells respond in vivo, where cells must integrate the signalling pathways activated by multiple stimuli.
One key proliferative signalling mediator is the extracellular signal-regulated kinase (ERK), a member of the mitogen-activation protein kinase (MAPK) family, and both LPA and EGF signal through ERK in many cell types 6, 7. However, ERK activation and transcription factors regulated by ERK are not the only mechanisms involved in proliferation. Numerous transcription factors are known to be activated downstream of both GPCRs and RTKs. Activator protein (AP)‐1, cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB), nuclear factor of activated T‐cells (NFAT), nuclear factor (NF)‐κB, the serum response element (SRE) complex and signal transducer and activator of transcription (STAT) are all known to be involved in proliferation and/or inflammation and have been implicated in the inflammatory state causing asthma and related airway diseases 8.
The synergistic activation of 3H thymidine incorporation by LPA plus EGF described previously 4, 5 implies that pathways activated by these two mitogens converge at some point in the nucleus or before. The point of convergence of the two pathways to mediate synergism represents a key regulatory step in the induction of mitogenesis. The purpose of this study was to identify sites for the signal integration involved in synergism and to utilise pharmacological inhibitors to investigate the mechanisms involved.
Material and methods
Study design
Because HASM cells exhibit synergistic stimulation of mitogenesis in response to LPA plus EGF, and since ERK is a key mediator of mitogenic signalling, the extent and duration of ERK activation were determined using Western blot analysis of cells treated with LPA, EGF and LPA+EGF. For the analysis of transcription factor activation as an alternative site of synergism, HASM cells were transduced with retroviral vectors containing a series of luciferase reporter genes driven by specific transcription factors potentially involved in proliferative signalling 9. During the course of these studies, the authors obtained evidence for involvement of the small guanosine 5′‐triphosphate (GTP)-binding protein Rho, and further studies of Rho activation were conducted using a Rhotekin Rho-binding domain peptide that selectively identifies the GTP‐bound activated state of Rho.
The luciferase activity of cells, transduced with reporter gene constructs for AP‐1, CREB, NFAT, NF‐κB, SRE complex and STAT, was assayed after treatment with LPA, EGF, or LPA+EGF. Pharmacological inhibitors of signalling pathways were used to assess the relative contributions of specific signalling pathways to the activation of each transcription factor and of ERK. Previously, a study investigating the times of mitogen exposure required for stimulation of deoxyribonucleic acid (DNA) synthesis revealed no synergism following an 8‐h exposure period to LPA+EGF, but strong synergism following a 12‐h exposure period 10. Therefore the current studies were focused on events that occurred during or prior to this time window.
Materials
Dulbecco's modified Eagle's medium (DMEM) and foetal bovine serum (FBS) were purchased from Life Technologies (Grand Island, NY, USA). 18:1 (oleoyl)‐LPA was purchased from Avanti (Alabaster, AL, USA) and reconstituted at 10 mM in 0.25% fatty acid-free bovine serum albumin from Sigma (St Louis, Mo, USA). Pertussis toxin (PTX) was purchased from List Biologicals (Campbell, CA, USA). AG1478, U0126, and rapamycin were purchased from Calbiochem (La Jolla, CA, USA). Calphostin C was obtained from Biomol (Plymouth Meeting, PA, USA), EGF was from Biosource International (Camarillo, CA, USA), and 3H thymidine was from NEN (Boston, MA, USA). Other chemicals were from Sigma.
C3 toxin preparation and cell treatment
The complementary DNA for a fusion protein of glutathione S‐transferase (GST) and Clostridium botulinum C3 toxin (pKG‐9E10.C3) was a generous gift of D. Alberts (Van Andel Research Institute, Grand Rapids, MI, USA). The vector was transformed into BL21 Escherichia coli (Stratagene, La Jolla, Ca, USA); C3 toxin was purified from logarithmically growing E. coli induced with 0.3 mM isopropylthiogalactoside. Cells were lysed in buffer containing 25 mM Tris pH 7.4, 100 mM NaCl, 10 mM MgCl2, 10 µg·mL−1 aprotinin, 1 mM phenylmethylsulphonyl fluoride and 1 mM dithiothreitol (DTT). Clarified extracts were incubated with glutathione-Sepharose (Molecular Dynamics/Amersham Pharmacia Biotech, Piscataway, NJ, USA). After washing, the GST tag was cleaved by incubation with thrombin overnight. Thrombin was removed by incubation with para-aminobenzamidine-Sepharose and C3 toxin was quantitated by Coomassie staining of protein resolved by sodium dodecysulphate polyacrylamide gel electropheresis (SDS‐PAGE). Cells were treated by adding the purified toxin to the culture medium, a method of delivery that has been shown to be effective in HASM cells previously 11.
HASM cell culture
HASM cells previously isolated from human trachea by enzymatic dissociation were kindly provided by M. Kotlikoff (Cornell University College of Veterinary Medicine, Ithaca, NY, USA) 12. HASM cells were cultured in high-glucose (4.5 g·L−1) DMEM with 10% FBS and passaged weekly. Human embryonic kidney (HEK)‐293T cells for retrovirus production were also grown in high-glucose DMEM with 5% FBS. Cells were routinely cultured at 37°C in a humidified 5% carbon dioxide incubator.
Extracellular signal-regulated kinase activation
HASM cells were plated on either 12‐well or 6‐well plates, grown to confluence and starved in serum-free DMEM for 24 h before being treated as indicated. After treatments the cells were lysed and lysates were loaded on 10% SDS‐polyacrylamide gels to separate the proteins. Proteins were transferred to membrane and blotted using an antiphospho‐ERK (Thr202/Tyr204) antibody, followed by an antirabbit horseradish peroxidase (HRP)‐conjugated secondary antibody (Cell Signaling Technology, Beverly, MA, USA). The signal was detected using the chemiluminescent LumiGLO reagent (Cell Signaling Technology) and exposure to film. Band density was quantitated using scanning laser densitometry and analysis with ImageQuant software (Molecular Dynamics/Amersham Pharmacia Biotech).
Production of retroviruses and transduction of HASM cells
Retroviral vectors based on the murine Moloney leukaemia virus and luciferase reporter genes had been generated previously to study transcription factor regulation in vascular smooth muscle cells 9. Each vector contained an enhancer for one of the following transcription factors, cloned upstream of a minimal interleukin (IL)‐2 promoter to drive regulated expression of luciferase: AP‐1, CREB, NFAT, NF‐κB, SRE complex (which includes Elk, ternary complex factor, serum response factor), and STAT. The pKA9 control vector contains the IL‐2 promoter and the luciferase gene but lacks any enhancer sequences. The amphotropic packaging vector pSV‐Ψ (-)‐A‐MLV, originally from O. Witte (University of California at Los Angeles, CA, USA), was obtained from T. Smithgall (University of Pittsburgh School of Medicine, PA, USA). Retroviruses were produced by cotransfecting the reporter gene-containing vector and the amphotropic packaging vector into HEK‐293T cells using a calcium phosphate transfection protocol. Retroviral supernatants were collected at 2, 3 and 4 days after transfection, filtered through 0.45 µm sterile filters and used immediately for transducing HASM cells. HASM cells passage four or five were plated at 40,000 cells·well−1 in 6‐well plates and transduced at ∼50% confluency. For transductions, 4 mL of retroviral supernatant and 0.8 mg·mL−1 polybrene was added to each well before cells were centrifuged for 30 min at 1,200×g in a Jouan 450 centrifuge (Jouan Laboratory Equipment, Winchester, VA, USA), at ∼32°C, essentially as described previously 13. Cells were centrifuged a total of four times with fresh retroviral supernatant each time. HASM cells were also transduced in parallel with a galactosidase-containing vector and stained with Xgal to determine transduction efficiency, which was >95%. After transduction, transduced cells were passaged and used in experiments for between three and six passages. HASM cells continued to grow and divide following transduction with the retroviral reporter constructs used in the authors' studies, in contrast to the cell cycle arrest previously reported with the use of adenovirus-mediated transduction of luciferase reporter constructs 14.
Luciferase assays
HASM cells were plated at 40,000 cells·well−1 in 6‐well plates and grown to confluence. Cells were then starved for 24 h in serum-free medium before treatment with mitogens in serum-free medium. Cells were then washed twice in Ca2+- and Mg2+-free phosphate-buffered saline and lysed in 200 µL lysis buffer containing 25 mM Tris, 4 mM ethyleneglycol-bis-(β‐aminoethylether)‐N,N,N′,N′‐tetraacetic acid (EGTA), 10% glycerol, 1% Triton X‐100, and 2 mM DTT. After a 10‐min incubation period in lysis buffer, cells were scraped and lysates were then clarified by centrifugation for 5 min at 20,000×g in a Beckman microcentrifuge (Beckman Coulter, Inc, Fullerton, CA, USA). Luciferase activity was assayed using a Pharmingen Monolight 3010 luminometer (Pharmingen, San Diego, CA, USA). 50 µL cell lysate was added to a cuvette containing 350 µL luciferase assay buffer (25 mM Tris, 20 mM MgSO4, 4 mM EGTA, 2 mM adenosine triphosphate and 1 mM DTT). After injection of 100 µL 1 mM luciferin in 10 mM DTT, relative light units were measured for 10 s following a 5‐s delay.
3H thymidine incorporation assays
The 3H thymidine assays were performed with confluent HASM cells starved in serum-free medium for 24 h and then treated with mitogens for 24 h, exactly as in previous studies 4.
Rho activation assays
Following the selective isolation of the active form of Rho, Western blotting was used to assess Rho activation; this method was based on the affinity for the Rhotekin Rho-binding domain. HASM cells were plated at 250,000 cells per 100-mm dish and grown to confluence. Cells were starved in serum-free medium for 24 h and then treated with mitogens for 3 min. Cells were washed twice with ice-cold Tris-buffered saline and then lysed in 0.5 mL Rho assay lysis buffer (50 mM Tris, pH 7.2, 500 mM NaCl, 10 mM MgCl2, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS, and 1 mM phenylmethylsulphonyl fluoride). The lysate was precleared by centrifugation for 2 min at 14,000×g. Activated Rho was then isolated using agarose beads conjugated to a GST‐fusion protein containing the Rho-binding domain of Rhotekin (Upstate Biotechnology, Lake Placid, NY, USA), which binds only the GTP‐bound active form of Rho. A 30‐µL aliquot of beads was added to the lysate from each dish, incubated with gentle rotation at 4°C for 45 min, isolated by centrifugation for 5 s at 14,000×g, and washed three times in Rho assay wash buffer (50 mM Tris, pH 7.2, 1% Triton X‐100, 150 mM NaCl, 10 mM MgCl2, and 0.1 mM phenylmethylsulphonyl fluoride). The beads were then resuspended in 20 µL of 2X Laemmli reducing sample buffer and boiled for 5 min. The beads were pelleted by centrifugation, and the supernatant was subjected to SDS‐PAGE on 10–20% gradient gels. Proteins were transferred to polyvinylidene difluoride membranes using a semi-dry transfer apparatus. These membranes were then blotted with mouse anti-RhoA antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), followed by incubation with HRP‐conjugated antimouse secondary antibody followed by visualisation and quantification as described above for ERK activation assays.
Analysis
Data were analysed using computer software. The stimulation values for the ERK and Rho activation experiments were calculated using densitometric analysis and were normalised by the conversion to fold stimulation over basal and then fold stimulation values for multiple experiments were averaged. The significance of the results was determined by using one-way analysis of variance (ANOVA) followed by the Bonferroni post-test.
For the transcription factor responses, data in each experiment were converted to fold stimulation over basal values and data from multiple experiments (using cells from two or three separate transductions) were then averaged. The one sample t‐test was used to determine the statistical significance of the activation of each luciferase reporter construct versus the control value of 1.0. The magnitude of the fold response was compared with the magnitude of the fold response in the enhancerless control vector pKA9. A value for stimulation that is not significantly different than the pKA9 control indicates that the stimulation is due to nonspecific transcriptional responses. The effect of treatment with LPA+EGF was compared with the sum of the treatments with LPA alone and EGF alone. Synergism was defined as stimulation of ≥150% of the sum of the stimulations by the two individual mitogens as stated in the authors' previous study 4. A two-way ANOVA was used to statistically compare the stimulation by LPA+EGF with the sum of the stimulations by LPA alone and EGF alone. In experiments testing inhibitors, the unpaired two-tailed t‐test was used to compare stimulation in the absence versus presence of the inhibitor.
Results
ERK activation in HASM cells treated with LPA, EGF and LPA+EGF
The extent and duration of ERK activation by LPA, EGF and LPA+EGF were determined during a 12‐h treatment period (fig. 1⇓), the time course in which synergism appears 10. LPA treatment of HASM cells yielded strong ERK activation, which was evident within 2 min, reaching a peak after 10 min. ERK activation by LPA was transient, with significant stimulation up to and inclusive of the 6‐h time point; after 6 h no significant ERK activation was observed. Treatment of HASM cells with EGF yielded similar ERK activation at the earlier time points; however, strong activation was retained over the entire time course, including 12 h post-treatment. When HASM cells were treated with LPA in combination with EGF, ERK activation paralleled that seen with EGF alone, showing strong activation throughout the time course (fig. 1⇓). Some of the experiments showed additive ERK activation responses at selected time points; the most common was at 6 h. However, this additive effect was not consistently present and a synergistic effect on ERK activation was not observed.

Extracellular signal-regulated kinase (ERK) activation was assessed in human airway smooth muscle cells after treatment with bovine serum albumin (BSA; ▪), 10μM lysophosphatidic acid (LPA; □), 60 ng·mL−1 epidermal growth factor (EGF; ○), or LPA+ EGF (•). The values shown represent the mean±sem of four to seven independent experiments. One-way analysis of the variance indicated ERK was significantly activated by LPA (p<0.05) up to and including the 6‐h time point, but not at later times. ERK activation by EGF and LPA+EGF was significant at all time points. The data shown in b) and d) indicate that the lower level of stimulation seen at 6 h primarily reflects an increase in the BSA control value. rdu: relative density units.
Signalling pathways involved in ERK activation by LPA and EGF
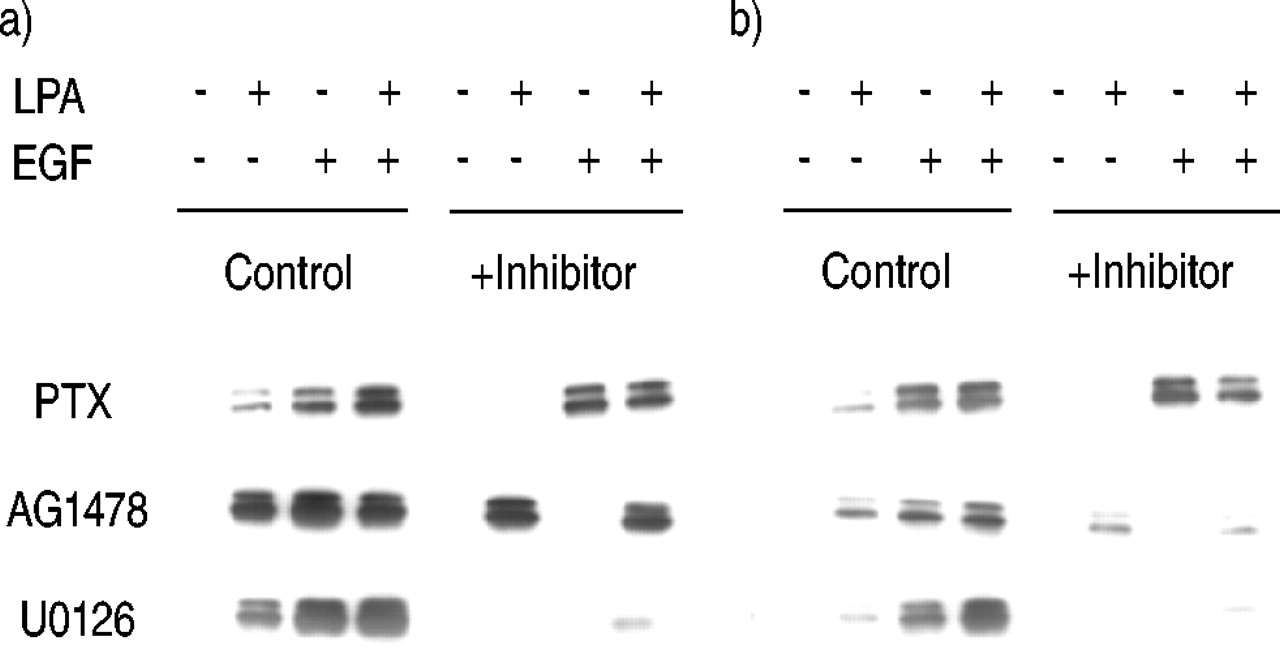
The signalling pathways leading to ERK activation by LPA, EGF and LPA+EGF were assessed using pharmacological inhibitors in either 10‐min or 2‐h assays. ERK activation by LPA was blocked by PTX pretreatment, implicating the inhibitory guanine nucleotide-binding protein (Gi) in ERK activation by LPA (fig. 2⇓). As expected, ERK activation by EGF was not inhibited by PTX. ERK activation by EGF was blocked by the EGF‐specific RTK inhibitor AG1478. In some systems GPCR mitogens, for example LPA, cause EGF‐independent transactivation of the EGF receptor (EGFR), therefore the EGFR is also thought to function in mitogenic signalling downstream of many GPCR mitogens 7, 15. However, AG1478 did not block ERK activation by LPA, suggesting that the EGF‐RTK is not required for LPA‐stimulated ERK activation. U0126 is an inhibitor of the MAPK‐ERK kinase (MEK), the kinase directly upstream of ERK and responsible for ERK activation. U0126 blocked ERK activation by both LPA and EGF, suggesting that ERK is activated primarily by MEK, although incomplete inhibition of the activation by LPA+EGF allows for the possible activation of ERK by an additional minor pathway. Densitometric analysis of multiple experiments revealed that U0126 inhibited ERK activation by LPA+EGF by only 76±4%, whereas U0126 blocked LPA‐stimulated ERK activation by 92±4% and EGF stimulated ERK activation by 99±1% (n=4).
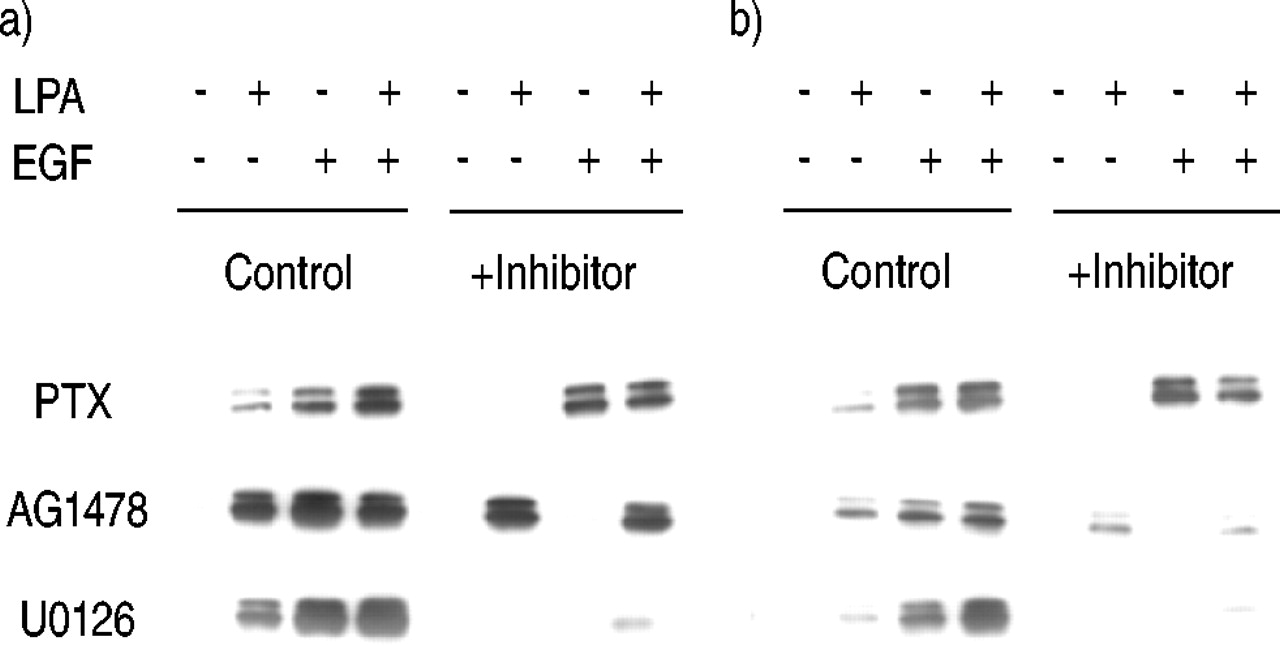
Effect of inhibitors on extracellular signal-regulated kinase (ERK) activation by lysophosphatidic acid (LPA) and epidermal growth factor (EGF) after a) 10‐min and b) 2‐h treatment. Human airway smooth muscle cells were treated with pertussis toxin (PTX; 100 ng·mL−1), AG1478 (2.5 µM), or U0126 (10 µM) for 30 min, then treated with bovine serum albumin, 10 µM LPA, 60 ng·mL−1 EGF, or LPA+EGF for either 10 min or 2 h, and phospho‐ERK was then analysed. A representative blot from at least three experiments is shown.
Other inhibitors were tested for their effects on ERK activation including the Src inhibitor PP1, the phosphatidyl inositol 3′‐kinase (PI3K) inhibitor LY294002, the ribosomal protein s6 kinase (p70s6k) inhibitor rapamycin, the phospholipase C inhibitor U73122, and the Rho inhibitor C3 toxin. None of these inhibitors were found to alter ERK activation in response to LPA, EGF, or LPA+EGF in either 10‐min or 2‐h assays (data not shown).
Activation of transcription factors after a 12‐h treatment with LPA, EGF, and LPA+EGF
HASM cells transduced with a panel of luciferase reporter constructs were treated with LPA, EGF, or LPA+ EGF for 12 h (table 1⇓). Cells transduced with the pKA9 vector, which does not contain an enhancer, were included as a negative control. Additionally, responses to prototypical agents for each transcription factor were included as positive controls.
Activation of transcription factor reporter gene constructs in human airway smooth muscle (HASM) cells
Treatment with LPA alone induced luciferase activity in cells bearing reporter gene constructs for AP‐1, CREB, NFAT, NF‐κB and the SRE complex. LPA did not significantly activate the STAT reporter construct. EGF induced transcription mediated by AP‐1, CREB, NFAT and the SRE complex. Although the values for NF‐κB and STAT were >1.0, they were not significantly greater than those for the control pKA9 vector, indicating that EGF does not specifically activate these transcription factors. When cells were treated with LPA+EGF, only the AP‐1 reporter construct exhibited synergistic activation. The reporter genes for CREB, NFAT and the SRE complex showed activation in the presence of both growth factors that was approximately the sum of those for the individual growth factors. In contrast, NF‐κB showed strong activation by LPA but not by EGF, and NF‐κB activation by LPA was not significantly altered by the presence of EGF.
Luciferase activity was also measured after 4 h of treatment. Interestingly, AP‐1 activation by LPA, EGF, and LPA+EGF was only additive at 4 h, in contrast to the synergistic response seen at 12 h. At 4 h the fold stimulation values were 2.4±0.4, 2.0±0.1, and 4.8±0.8, respectively (n=3). Values for stimulation of the other reporter constructs were similar at both 4 and 12 h; NF‐κB activation by LPA at 4 h was 4.7±1.2‐fold (n=4) and again no significant activation of NF‐κB by EGF was observed (1.3±0.1 fold; n=4).
Signalling pathways involved in AP-1 activation by LPA and EGF
Because the transcription factor AP‐1 is well established as a critical mediator of proliferation 16 and was synergistically activated by LPA+EGF, the effects of signalling pathway inhibitors on AP‐1 activation by LPA and EGF were investigated (fig. 3⇓). Pretreatment of cells with PTX completely blocked LPA‐mediated AP‐1 activation, implicating Gi in LPA‐stimulated AP‐1 activation. PTX caused only a small (but statistically significant) inhibition of AP‐1 activation by EGF. In the presence of PTX, activation of AP‐1 by LPA+EGF was equal to that seen with EGF alone. Because LPA can also activate Gq and phospholipase C in HASM cells 17, a possible role of protein kinase C (PKC) in AP‐1 activation was investigated. The PKC inhibitor calphostin C did not block AP‐1 activation by LPA, EGF, or LPA+EGF (fig. 3⇓), suggesting that PKC activation is not required for AP‐1 activation by any of these stimuli.
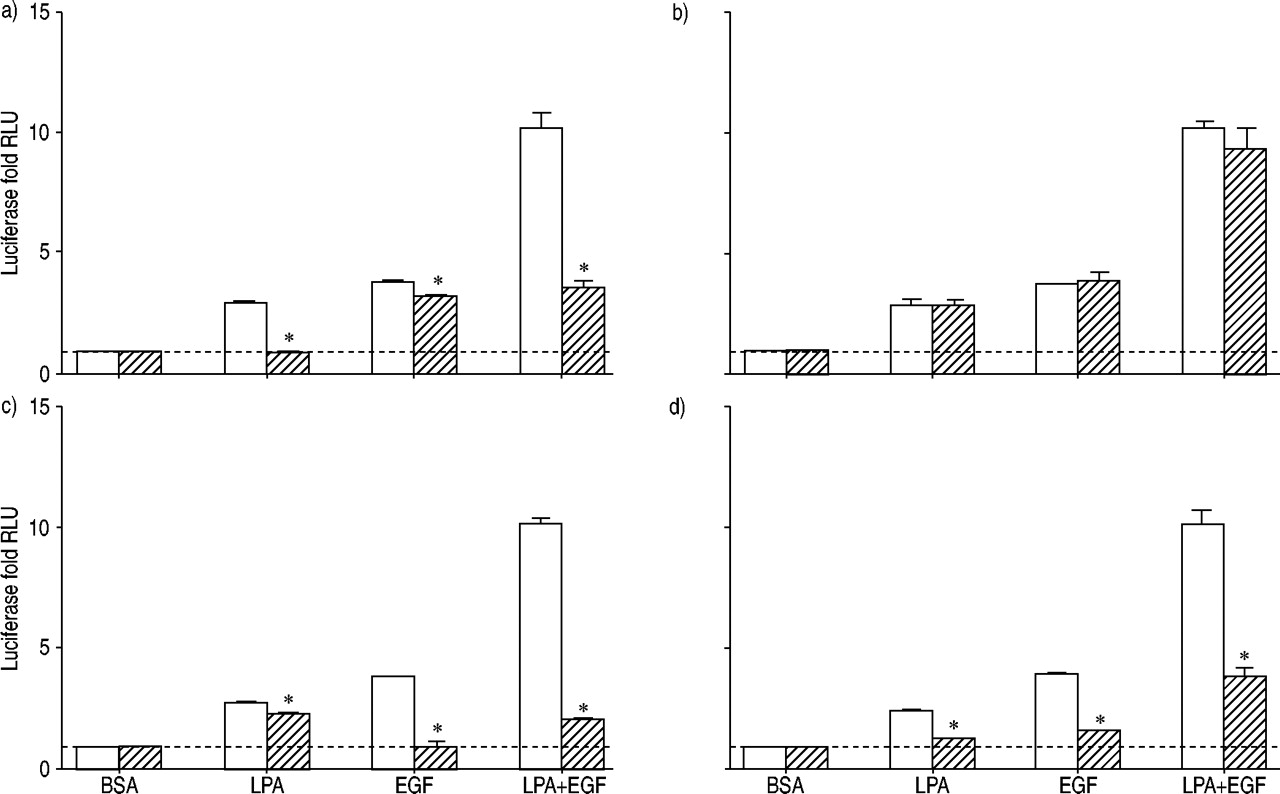
Luciferase activity was assessed for human airway smooth muscle cells containing the activator protein (AP)‐1 reporter gene, treated for 12 h with 10 µM lysophosphatidic acid (LPA), 60 ng·mL−1 epidermal growth factor (EGF), LPA+EGF, or the appropriate bovine serum albumin (BSA) control in the absence (□) or presence (└) of inhibitors: a) pertussis toxin 100 ng·mL−1, b) calphostin C 1 µM, c) AG1478 2.5 µM, and d) U0126 10 µg·mL−1. Values shown represent the mean±sem for three to six experiments using cells from two transductions. ---: baseline as normalised to BSA control; RLU: relative light units. *: p<0.05.
Treatment of cells with LPA plus the EGF‐RTK inhibitor AG1478 did not block LPA‐stimulated AP‐1 activation (fig. 3⇑), although a small but statistically significant inhibition was observed. These results suggest that the tyrosine kinase activity of the EGFR plays a minor role, if any, in mediating LPA‐stimulated AP‐1 activation. AG1478 did block EGF‐mediated AP‐1 activation, as expected. Stimulation with LPA+EGF in the presence of AG1478 was the same as that seen with LPA alone. Incubation of cells with both PTX and AG1478, to block Gi‐mediated LPA effects and EGFR‐mediated EGF effects, completely blocked AP‐1 activation by LPA+EGF, as expected (data not shown).
The MEK inhibitor U0126 blocked AP‐1 activation by LPA and EGF, implicating ERK in the activation of AP‐1 by both agents (fig. 3⇑). Interestingly, however, U0126 did not completely block AP‐1 activation by the combination of LPA+EGF. In the absence of U0126, AP‐1 activation by LPA+EGF was increased by 10.2±1.1‐fold over basal, whereas in the presence of U0126, AP‐1 activation by LPA+EGF was still increased by 3.5±0.1‐fold. The inability of U0126 to completely block AP‐1 activation by LPA+EGF is consistent with the observation that U0126 does not completely block ERK activation by LPA+EGF (fig. 2⇑). This suggests that the failure of U0126 to completely block AP‐1 activation by LPA+EGF may be a result of its incomplete inhibition of ERK activation by this combination of mitogens. Alternatively, there may be an ERK‐independent component of AP‐1 activation by LPA+EGF.
A recent study suggested a role for p70s6k in mitogenic signalling and synergism in HASM cells 14. However, in the current experiments, the p70s6k pathway inhibitor rapamycin did not inhibit AP‐1 activation and may have enhanced AP‐1 activation. The fold stimulation values for LPA, EGF, and LPA+EGF were 2.5±0.2, 3.3±0.3 and 9.3±0.1 in the absence of rapamycin and 3.2±0.2, 4.8±0.3 and 16.3±0.6 in the presence of 1 µM rapamycin (n=3). Therefore, p70s6k does not appear to be required for synergistic AP‐1 activation in HASM cells and any contribution of p70s6k to mitogenesis or synergism in these cells is likely to be through mechanisms other than AP‐1 activation.
Involvement of ERK and Gi activation in LPA and EGF regulation of other transcription factors
Signalling pathways for LPA‐mediated activation of transcription factors other than AP‐1 were investigated. The effects of U0126 on activation of CREB, the SRE complex, NFAT and NF‐κB by LPA, EGF and LPA+EGF were assessed (fig. 4⇓). U0126 did not significantly inhibit LPA stimulation of CREB activation; however, stimulation by EGF was markedly inhibited. CREB activation by LPA+EGF was not significantly inhibited. Results for the SRE complex were similar to those for CREB; i.e. LPA was weakly inhibited, EGF was markedly inhibited and LPA+EGF was marginally decreased. U0126 almost completely blocked NFAT activation by LPA, EGF and the combination of LPA+EGF. In contrast, U0126 had no effect on NF‐κB activation by LPA or the combination of LPA+EGF.

Luciferase activity was assessed in human airway smooth muscle cells containing a) the cyclic adenosine monophosphate response element-binding protein, b) serum response element, c) nuclear factor of activated T‐cells, and d) nuclear factor‐κB reporter genes, treated for 12 h with 10 µM lysophosphatidic acid (LPA), 60 ng·mL−1 epidermal growth factor (EGF), LPA+ EGF, or the appropriate bovine serum albumin (BSA) control in the absence (□) or presence (└) of 10 µM U0126. Values shown represent the mean±sem of three to four experiments with two cell transductions. ---: baseline as normalised to BSA control; RLU: relative light units. *: p<0.05.
The effects of PTX on LPA‐mediated activation of CREB, the SRE complex, NFAT, and NF‐κB were assessed (fig. 5⇓). Activation of CREB and NFAT were almost completely blocked by PTX. Activation of the SRE complex was largely PTX‐sensitive, whereas NF‐κB activation by LPA was only modestly reduced by PTX. EGF activation of all of these responses was PTX‐insensitive, as expected. For the SRE complex, CREB and NFAT, the PTX‐treated cells exhibited responses to LPA+EGF similar to those seen with EGF alone.

Luciferase activity was assessed in human airway smooth muscle cells containing a) the cyclic adenosine monophosphate response element-binding protein, b) serum response element, c) nuclear factor of activated T‐cells, and d) nuclear factor‐κB reporter genes, treated for 12 h with 10 µM lysophosphatidic acid (LPA), 60 ng·mL−1 epidermal growth factor (EGF), LPA+EGF, or the appropriate bovine serum albumin (BSA) control in the absence (□) or presence (└) of 100 ng·mL−1 pertussis toxin. Values shown represent the mean±sem of three to four experiments with two cell transductions. ---: baseline as normalised to BSA control; RLU: relative light units.*: p<0.05 using a two-tailed t‐test.
The effects of the Rho‐inactivating C3 toxin on LPA activation of NF‐κB and AP‐1 and synergistic stimulation of mitogenesis
The relative insensitivity of LPA‐mediated NF‐κB activation to either PTX or U0126 suggested that an alternate pathway to NF‐κB activation might be involved. Recent reports have proposed Rho as a mediator of NF‐κB activation by other GPCR agonists 18, 19. Accordingly, the effects of the Rho inhibitor C3 toxin on NF‐κB activation were tested. C3 toxin eliminated NF‐κB activation by LPA, implicating Rho in NF‐κB activation (fig. 6⇓). Because C3 toxin blocked NF‐κB activation and because a component of AP‐1 activation by LPA+EGF was insensitive to U0126, the effect of C3 toxin on AP‐1 activation was also tested. Treatment of cells with C3 toxin did not block the stimulation of AP‐1 by either LPA or EGF. However, C3 toxin eliminated the synergism in AP‐1 activation by LPA+EGF. These results suggest that Rho may be uniquely involved in the synergistic activation of AP‐1 by LPA+EGF, even though it does not appear to be required for a response to either growth factor alone.

Luciferase activity was assessed in human airway smooth muscle cells containing either a) nuclear factor (NF)‐κB or b) the activator protein (AP)‐1, treated for 12 h with 10 µM lysophosphatidic acid (LPA), 60 ng·mL−1 epidermal growth factor (EGF), LPA+EGF, or the bovine serum albumin (BSA) control in the absence (□) or presence (└) of 10 ng·mL−1 C3 toxin. Values are presented as the mean±sem of two to four experiments using two cell transductions. C3 toxin increased basal values to 2.9±0.4‐fold of control for AP‐1 cells, 7.8±1.3‐fold of control for NF‐κB cells, and 1.7±0.1‐fold of control for serum response element complex cells. The values shown are the fold increases for treated cells relative to these elevated control values. ---: baseline as normalised to BSA control; RLU: relative light units. *: p<0.05 using a two-tailed t‐test.
The effect of C3 toxin on mitogenesis assessed by 3H thymidine incorporation into DNA was also investigated. It was found that C3 toxin partially inhibited mitogenesis stimulated by LPA alone; however, it had no effect on stimulation by EGF alone (fig. 7⇓). Importantly, synergism was eliminated by C3 toxin, with stimulation by LPA+EGF reduced to levels that were only additive rather than synergistic. This suggests that Rho activation is involved in the synergism between LPA and EGF at the level of DNA synthesis, similar to the results obtained above with AP‐1 activation assays (fig. 6⇑).

3H thymidine incorporation was assessed for human airway smooth muscle cells treated with bovine serum albumin (BSA), 10 µM lysophosphatidic acid (LPA), 60 ng·mL−1 epidermal growth factor (EGF), or LPA+ EGF for 24 h in the absence (□) or presence (└) of 10 µg·mL−1 C3 toxin. Values shown represent the mean±sem for three experiments. ---: baseline as normalised to BSA control; *: p<0.05 using a two-tailed t‐test.
Effects of LPA and EGF on Rho activation
Because of the inhibitory effect of C3 toxin specifically on the synergistic component of HASM cell mitogenesis, the effects of LPA, EGF and LPA+EGF on Rho activation were assessed. These studies were undertaken to confirm that Rho is activated, to identify which mitogen mediates Rho activation and to determine whether synergism is observed at the level of Rho activation. The Rho-binding domain of Rhotekin attached to agarose beads as a GST‐fusion protein was used to isolate the GTP‐bound active form of Rho, which was then quantified by immunoblotting with anti-Rho antibodies (fig. 8⇓). LPA induced a marked stimulation of Rho activation, but there was no significant activation of Rho by EGF. The combination of LPA+EGF did not exhibit synergism in Rho activation but instead appeared less effective than LPA alone.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rho activation was assessed for human airway smooth muscle cells stimulated with bovine serum albumin (BSA), 10 µM lysophosphatidic acid (LPA), 60 ng·mL−1 epidermal growth factor (EGF), or LPA+EGF for 3 min. Values are expressed as the fold stimulation compared with the BSA control and are the mean±sem from three experiments. ---: baseline as normalised to BSA control; *: p<0.05 by one-way analysis of variance followed by the Bonferroni post-test.
Discussion
The aim of these studies was to investigate potential points for integration of LPA- and EGF-signalling pathways downstream of receptor activation in HASM cells and to identify the point at which these pathways converge to mediate synergistic stimulation of DNA synthesis. Both LPA- and EGF-activated pathways led to ERK activation; however, synergism was not observed at the level of ERK activation. Both LPA and EGF led to the activation of multiple transcription factors including CREB, NFAT, AP‐1, and the SRE complex. Furthermore, the ERK signalling pathway appeared to be involved in activating these transcription factors based on the inhibitory effects of U0126. Treatment of HASM cells with LPA+EGF generated synergistic activation of AP‐1. Inhibition of Rho activation with C3 toxin blocked the synergism in AP‐1 activation and synergistic mitogenesis, implicating Rho in synergistic signalling. LPA rather than EGF appeared to be the primary factor mediating this Rho activation. Therefore, both ERK activation and Rho activation are required for mitogenic signalling; however, neither is the locus of synergism. AP‐1 activation does exhibit synergism and as such represents a likely point of integration for signals from LPA and EGF.
The signalling pathways to ERK activation were investigated because of the likely importance of ERK activation in leading to synergism at some downstream locus. Consistent with the known pathways for ERK activation in other cell types, activation by EGF required the tyrosine kinase activity of the EGFR. Activation by LPA was mediated by Gi or another PTX‐sensitive G‐protein but did not appear to require EGF‐RTK activity. This finding is consistent with another recent study from the authors' laboratory that showed more directly that LPA does not stimulate tyrosine phosphorylation of the EGFR 10. MEK activation was required for ERK activation by both LPA and EGF.
Several lines of evidence indicate that ERK activation is not sufficient for synergism. First, ERK activation by LPA+EGF was no greater than by EGF alone. Second, although sustained ERK activation appeared to be critical for signalling proliferation in several cell types including HASM cells 20–22, activation of ERK by LPA was transient with insignificant ERK activation by LPA between the 8–12 h treatment period, which is the key time-window for synergism 10. However, ERK activation by EGF was sustained through a 12‐h period of exposure showing similar results to a previous study with HASM cells 21. This sustained ERK activation may represent the major contribution by EGF to synergistic mitogenic signalling in HASM cells. Third, PTX sensitivity of ERK activation by LPA argues against ERK as the key to synergism, since synergism between LPA and EGF is not PTX‐sensitive 5. Although ERK activation itself is not synergistic, activation of ERK is nonetheless critical for both mitogenic signalling and for synergism, based on the inhibitory effects of U0126 on transcription factor responses downstream of ERK activation.
In a recent study, the authors showed that treatment of HASM cells with LPA induced an approximate two-fold upregulation of EGFR‐binding activity that was mediated by increased expression of EGFRs 10. Upregulation of EGFRs by LPA was an attractive mechanism for explaining synergism, since it could explain the requirements for both LPA and EGF for maximal stimulation and because the time course for EGFR upregulation correlated with the time course for the onset of synergism. An obvious prediction from this model was that the upregulation of EGFRs would lead to a corresponding increase in EGF‐stimulated ERK activation. However, in the current studies ERK activation was not greater with LPA+EGF than with EGF alone at 12 h, a time point at which maximal EGFR upregulation by LPA had occurred 10. These results suggest that any contribution of EGFR upregulation to synergism occurs through a signalling pathway other than ERK activation.
The current studies also document activation of multiple transcription factors by both LPA and EGF in HASM cells. To the best of the authors' knowledge, this is the first report of LPA‐mediated CREB activation. Although LPA has inhibitory effects on cAMP formation, CREB can also be activated through ERK, PI3K, and calcium-signalling pathways 23, all of which are activated by LPA in these or other cells 6. Similarly, LPA‐mediated NFAT responses have not been documented previously. NFAT is primarily regulated through calcium signalling and calcineurin activation, and LPA does increase intracellular calcium in HASM cells (unpublished data). NF‐κB activation by LPA has been demonstrated for fibroblasts and endothelial cells 24, 25. The effect of LPA to increase SRE complex activation is well established for fibroblast cell lines 26. Since c‐fos is a component of AP‐1 and the c‐fos gene contains well-characterised SRE and CRE sequences 27, transcriptional increases in c‐fos expression by activation of these factors is a potential explanation for the ability of LPA to activate AP‐1, although this remains to be established. EGF has been demonstrated to activate the c‐fos SRE 26 and CREB 28 in other cell types and this may also be the mechanism for EGF activation of AP‐1. Although EGF is not a classical activator of NFAT, a recent report suggests that EGF may activate NFAT through activation of Vav1 in T‐cells 29.
All EGF‐mediated responses were inhibited by AG1478, indicating mediation by the classical EGF‐RTK pathway. All EGF responses were also blocked by U0126, implicating MEK and presumably ERK as downstream-signalling mediators of each of these transcription factor responses to EGF. AP‐1, CREB, SRE and NFAT responses to LPA were also inhibited by U0126. However, activation of the NF‐κB reporter gene was completely insensitive to U0126. Similarly, LPA activation of AP‐1, CREB, SRE and NFAT was PTX‐sensitive, whereas NF‐κB activation was not PTX‐sensitive.
Rho is a small G‐protein that is best characterised as a modulator of cytoskeletal responses, but recent evidence clearly implicates Rho in regulating gene transcription and cell proliferation 30. Importantly, the Rho inhibitor C3 toxin blocked synergistic AP‐1 activation by LPA+EGF but not AP‐1 activation by either LPA or EGF alone. Therefore, Rho activation appears to be required for the additional AP‐1 activation observed when LPA and EGF are present simultaneously. These data implicate Rho as a key component of the signalling pathway leading to synergism. Results with C3 toxin in HASM cell mitogenesis assays suggest that Rho activation may be a key to the synergism observed in the mitogenesis assessed by 3H thymidine incorporation, which is also similar to the results for the activation of AP‐1. These studies point to a role for Rho as a key signal for synergistic signalling in proliferation. Further studies indicated that LPA rather than EGF is the primary mediator of Rho activation, consistent with recent evidence for LPA activation of Rho in HASM cells 31 and also in other cell types 32. The mechanisms involved in LPA activation of Rho in these cells and the relevant Rho effectors involved in synergism are exciting directions for future research.
NF‐κB activation was minimally sensitive to PTX and not affected by U0126, suggesting that NF‐κB activation occurs by an alternate LPA‐signalling pathway. Because synergism is not PTX‐sensitive and not mediated by ERK, this alternative NF‐κB pathway could also contribute to synergism. Interestingly, both NF‐κB and Rho were activated by LPA but not by EGF. Furthermore, NF‐κB activation by LPA was blocked by C3 toxin, suggesting that Rho activation is required for LPA to activate NF‐κB. This result is consistent with other recent reports that NF‐κB activation may be regulated by Rho. Rho-mediated NF‐κB activation has been demonstrated for bradykinin treatment of A549 lung carcinoma cells 18 and sphingosine‐1‐phosphate treatment of HEK293 cells 19. Thus, Rho-mediated NF‐κB activation is an intriguing possibility for the contribution of LPA to the synergism observed in both AP‐1 activation and mitogenesis assays.
A central role for AP‐1 is supported by a recent study investigating other mitogen combinations that are synergistic in HASM cells, specifically EGF plus thrombin, histamine, and carbachol 14. Although ERK activation by these combinations was not synergistic, enhanced activation of both AP‐1 and Elk‐1 was observed, even though the use of adenovirus-mediated transduction of the luciferase reporter constructs caused cell cycle arrest 14. The time course data from the present study were also consistent with a role for AP‐1 in synergism, as AP‐1 activation was not synergistic after 4 h of treatment with LPA+EGF but was after 12 h of treatment. These results parallel the previously reported time course for synergistic activation of mitogenesis, where 12 h of treatment with LPA+EGF was required to observe synergism 10. Further studies of AP‐1 activation to characterise the specific AP‐1 factors involved in synergism and how they are regulated by signalling pathways activated by LPA and EGF may provide further insight into the mechanisms of AP‐1 synergism. Additionally, activation of AP‐1 could further synergise with other transcriptional factors such as NF‐κB by interacting directly at the promoter sites of genes that play critical roles in regulating proliferation. Since AP‐1 and NF‐κB have both been implicated in the regulation of inflammatory signalling that occurs in diseases of airway remodelling, such as asthma, further studies of the regulation of these two transcription factors and their activation should yield results relevant to the understanding of the pathogenesis and treatment of airway diseases.
A recently published study suggests that the simultaneous activation of Ras-mediated and Rho-mediated pathways can mediate synergism in vascular smooth muscle cells. Constitutively active Ras and Rho constructs transfected together into rat vascular smooth muscle cells caused synergistic stimulation of proliferation 33. Based on the results in the current study, a similar model is proposed for airway smooth muscle cells, with Ras-activated and Rho-activated pathways interacting to cause synergism 3. EGF is proposed to contribute to the synergism through sustained ERK activation, presumably downstream of Ras. This sustained ERK activation is not seen in cells treated with LPA alone. LPA contributes to synergism through activation of a Rho-mediated pathway, with one candidate for a downstream effector of Rho in this system being NF‐κB. Thus synergistic activation of AP‐1 and the synergistic stimulation of mitogenesis in HASM cells may result from the effect of LPA to activate Rho together with the strong effect of EGF to activate Ras and to mediate a sustained activation of ERK. The convergence of signalling pathways activated by Ras and Rho to modulate transcription factors including NF‐κB and AP‐1 presents a simple and elegant model for synergistic signalling to mitogenesis by LPA and EGF activation of interacting small G‐protein pathways.
These studies identify many of the important transcription factors stimulated by lysophosphatidic acid and epidermal growth factor and key signalling pathways that regulate their activity in airway smooth muscle cells. More detailed investigations of the molecular mechanisms involved in the interactions between these mitogenic-signalling pathways may provide new targets for modulating the increase in airway smooth muscle seen in vivo in the pathological conditions of asthma and other diseases involving airway remodelling.
Acknowledgments
The authors would like to thank J. Lynch for assistance with the statistical analyses, T. Smithgall, O. Witte, A. Alberts and M. Kotlikoff for reagents, and R. Lewis for helpful discussions.
- Received August 15, 2002.
- Accepted December 13, 2002.
- © ERS Journals Ltd