



Comparative mycobacterial genomics as a tool for drug target and antigen discovery
- S.T. Cole
- S.T. Cole, Unité de Génétique Moléculaire Bactérienne, Institut Pasteur, 28 rue du Docteur Roux, 75724 Paris Cedex 15, France. Fax: 33 140613583. E‐mail: stcole@pasteur.fr
Abstract
Genomics and the associated downstream technologies are generating vast data sets that provide new opportunities for understanding and combating both infectious and genetic diseases in humans.
The genomic approach has been applied to tuberculosis, a major cause of transmissible morbidity and mortality, with notable success. Complete genome sequences are now available for three members of the Mycobacterium tuberculosis complex and the related intracellular pathogen M. leprae.
Many of the predictions generated in silico by genomics have been validated through functional analysis, including studies of the transcriptome and proteome, and led to the identification of essential genes. Knowledge of the latter defines potential targets for new and existing drugs and their specificity can be assessed by comparative genomics with the host or other pathogens. Genomics is also furthering tuberculosis vaccine development by pinpointing potentially antigenic proteins as well as providing better diagnostic tools to detect infection.
- drug targets
- functional genomics
- genomics
- leprosy
- tuberculosis
There is an ever-growing need for new drugs and vaccines to treat and prevent mycobacterial diseases, particularly tuberculosis (TB), and for improved diagnostic tools to detect infection more reliably. The 1990s saw the widespread emergence of multidrug-resistant strains of Mycobacterium tuberculosis (MDR-TB) in both the developing and industrialised nations 1, 2. Transmission of MDR-TB has been documented, particularly among the human immunodeficiency virus (HIV)-infected, and the spectre of untreatable disease is fast becoming a reality. Although it is widely accepted that vaccination is the most desirable means of preventing TB, there is extensive evidence indicating that the current vaccine, bacille Calmette-Guérin (BCG), is only effective against the rarer, disseminated forms of the disease 3, 4. BCG has very limited efficacy against pulmonary TB that accounts for most of the disease burden 5 and has consistently failed to confer significant protection in developing countries despite inducing protective responses against leprosy in the same settings 6. The latter observation indicates that the leprosy bacillus probably shares common antigens with BCG and, in turn, with its close relative M. tuberculosis.
Tuberculosis drug development: ancient and modern
Most of the antituberculous drugs in current use were developed in the 1950s and arose from microbiological screens of libraries of natural or chemical compounds, or as the result of serendipitous leads. In 1943, streptomycin, the first antibiotic discovered, was shown to cure infections due to some Gram negative bacteria, and shortly after to be highly potent against M. tuberculosis. The chance observation 7 that nicotinamide inhibited the growth of mycobacteria in mice led to the synthesis and testing of related compounds, and culminated in the exquisitely specific drug isoniazid (INH) 8. Activity testing was initially performed on in vitro grown organisms and then confirmed in an animal model of infection before clinical trials were undertaken with promising molecules. Subsequently, additional drugs were developed that inhibited particular features of M. tuberculosis, such as the biogenesis of its remarkable cell wall. Ideally, antibacterial agents display bactericidal activity and target essential activities. One means of pinpointing such functions, which has never been applied to the tubercle bacillus, is to isolate and characterise mutants with conditionally lethal defects and then to screen for inhibitors capable of generating the same effect. This could be done by monitoring microbiological parameters, such as growth rate, or by using an in vitro assay if suitable functional tests exist. Nowadays, identification of new drugs can result from a rational, hypothesis-driven approach inspired by genomics or from high-throughput screening of chemical or combinatorial libraries by a variety of automated methods. The desired properties of new antitubercular agents include reduction of the duration of treatment, as well as activity against latent TB infections and MDR-TB strains 9.
Genomics
Genomics, the systematic study of the complete set of genetic material in the cell, through deoxyribonucleic acid (DNA) sequencing and bioinformatic analysis, offers vast potential in terms of drug target and antigen discovery and is enhancing the development of new antibacterial agents and vaccines. For a relatively modest, single investment the entire complement of genes present within a pathogen can be defined and their sequences compared with those in the genome sequences of other organisms including microbes, mice and men 10, 11. In the field of TB research, genomics was first applied to H37Rv 12, the widely used paradigm strain of M. tuberculosis, and then later to CDC1551 13, a recent clinical isolate from the USA, and then to the AF2122/97 strain of M. bovis 14, which was responsible for TB epidemics among cattle and badgers in the UK (table 1⇓). The genome sequence of a close relative of the tubercle bacillus, M. leprae, has also been determined 15.
Websites with information pertinent to genomics transcriptomics, proteomics and structrual of Mycobacterium tuberculosis
Genomics and drug target discovery
The genomes of all three fully-sequenced members of the M. tuberculosis complex contain ∼4.4 (+/− 0.1) Mb and harbour ∼4,000 genes that are predicted to encode proteins and 50 genes for stable ribonucleic acid (RNA) species. In the case of M. tuberculosis H37Rv, bioinformatic analysis resulted in the attribution of precise functions to ∼40% of the 4,000 genes. Some functional information was inferred for a further 20%, but nothing was learned about the remaining 40% 12, 16. When functional information was available, it often enabled investigators to identify potential drug targets on the basis of their proposed biological role or their similarity to known bacterial drug targets. However, now that more mycobacterial sequences are becoming available, it is possible to establish which genes are generally found in mycobacteria or restricted to a given species 17, 18. The functions encoded by these genes, if essential, could represent novel targets for chemotherapy that are exceptionally specific. In contrast, a large number of genes of unknown function have been found in many bacteria and these are commonly known as the conserved hypothetical genes 19, 20. Their widespread conservation is certainly of biological significance and some of these genes were subsequently found to play critical roles. Thus, they represent novel targets for new broad spectrum antibiotics 19. To ensure that new antibiotics inhibit functions that are confined to bacteria, thereby reducing potential side-effects in humans, it is now possible to perform in silico screening of the human genome sequence 10, 11 to exclude the possibility that related genes or proteins will be found in the host. Similar screens of the genome sequences of other pathogens can be undertaken to enhance specificity. Among the many attractive features of highly specific drugs, like INH or pyrazinamide, are the avoidance of transferable drug resistance mechanisms, such as those that have plagued certain broad spectrum antibiotics, and the reduction of unwanted side-effects, like the indiscriminate destruction of the bowel flora.
Validating drug targets through functional genomics
Several different approaches are available to determine which genes of M. tuberculosis are essential and thus worthy of further investigation as targets for drug development. These include gene knockouts, transcript analysis and definition of the proteome. Following is a brief description of the strategies available. Having identified potential candidates, it is important to demonstrate that the genes are expressed, particularly during infection, and here transcriptomics and proteomics offer great promise by allowing global analyses to be undertaken. All of these approaches are considerably facilitated by the availability of the complete genome sequence 12.
Transcriptomics
Various methods have been used to monitor the transcriptome, the complete set of RNA molecules produced by tubercle bacilli, including both targeted and random approaches. Among the latter are differential expression of customised amplification libraries (DECAL) 21 and selective capture of transcribed sequences (SCOTS) 22, techniques that combine polymerase chain reaction (PCR) and subtractive hybridisation in order to identify genes that are expressed differentially. When DECAL was applied to detect differences in expression in response to treatment with INH, three overexpressed genes (iniA, iniB and iniC) whose expression was also induced by ethambutol, another drug targeting the cell envelope, and by other treatments that influence cell wall functions, were found 21. This suggested that the role of iniABC may be pivotal in cell envelope biogenesis, although its function remains unknown.
On application of SCOTS to detect genes differentially expressed in human primary macrophages 22, it was established that several genes were upregulated. Among these were two alternative sigma factors (sigE and sigH) that had been previously implicated in stress survival 23, a polyketide synthase (pks2), isocitrate lyase (aceA also known as icl), an enzyme that has been shown to be required for long-term persistence of M. tuberculosis in infected mice 24, and mce1B, a gene locus that has been shown to confer the ability to invade and survive within HeLa cells to Escherichia coli 25. Of these candidates, isocitrate lyase, whose crystal structure is available 26, appears to be the most promising drug target as it intervenes in the glyoxylate shunt, a biochemical pathway confined to certain microbes and plants.
DNA microarrays are a powerful tool for studying differential gene expression and typically consist of gene-specific probes, immobilised on a solid surface such as glass, which serve as hybridisation templates 27. Microarrays can be used to compare gene expression in vivo with that observed in vitro, although results from M. tuberculosis growing in tissue have not yet been reported. However, in a most elegant study, Wilson et al. 28 examined the transcriptional response of the tubercle bacillus to INH. Overexpression of genes encoding components of the FAS-II fatty acid synthase system was detected, as expected from findings of proteomics 29, together with fbpC, which encodes the abundantly secreted antigen 85‐C that has trehalose-dimycolyl transferase activity and intervenes in the final steps of mycobacterial cell wall synthesis 30. Other INH-induced genes were fadE23 and fadE24, encoding two acyl-coenzyme A dehydrogenases involved in the β‐oxidation of fatty acids, and ahpC, encoding alkyl-hydroperoxide reductase, which may respond to a secondary toxic effect of INH. Two other sets of upregulated genes deserve further comment: efpA, encoding a putative efflux system that may play a role in innate drug resistance 28 and iniAB, which were identified by DECAL 21 and are discussed above. As can be seen from these studies, microarrays and related transcriptome tools are extremely useful for identifying coregulated genes and could help pinpoint sets of genes and proteins that act cooperatively. This represents a powerful means of uncovering additional drug targets within the same pathway, thereby enhancing the chances of synergistic effects.
Proteomics
Another means of monitoring gene expression is by studying the proteome, the complete set of proteins produced by the tubercle bacilli under different conditions. The most thorough study of the proteome of different strains of the M. tuberculosis complex was reported by Jungblut et al. 31, who used a combination of two-dimensional electrophoresis and mass spectrometry to resolve and identify proteins, respectively. Similar approaches have been applied to culture filtrate 32 and soluble proteins by Andersen and coworkers 33, 34. Details of websites providing this information are given in table 1⇑.
Comparative proteomics was undertaken with two different M. tuberculosis strains (H37Rv and Erdman) and two strains of M. bovis BCG (Copenhagen and Chicago). One-thousand eight-hundred spots corresponding to mycobacterial cell proteins and >200 in-culture supernatants were detected, of which 263 and 54 were identified, respectively 31, 35. Of the identified proteins, some perform housekeeping functions while others participate in the metabolism of fatty acids and glycolipids. Heat shock proteins were particularly abundant, whereas only three spots correspond to putative cell envelope proteins.
Proteomics can also be used as a tool for antigen discovery. Comparisons between BCG Chicago and H37Rv revealed 31 variable proteins. Thirteen were present in H37Rv, but not in the vaccine strain, six of which were identified. Eight proteins appeared to be absent from H37Rv, nine were downregulated, and one was overexpressed with respect to BCG. Eight of the differences were due to mobility variants, possibly caused by amino acid changes or post-translational modifications. Further comparisons between two M. tuberculosis strains revealed only minor differences in protein expression profiles in vitro 31. This finding has been confirmed in an independent study in which the proteomes of the strains H37Rv and CDC1551 were compared 36.
More recently, two-dimensional liquid-phase electrophoresis has been employed to obtain several hundred fractions of culture filtrate and cytosolic proteins, respectively. These were screened for their ability to stimulate T‐cell responses and 30 individual proteins were identified by mass spectrometry, 17 of which were novel T‐cell antigens 37. At present there is little information available about the proteomes of tubercle bacilli isolated from macrophages, or even diseased tissue, and this should be investigated intensively in the coming years. Similarly, little is known about proteins present in the particulate fraction of the cell and, when one considers that 60% of known drug targets are membrane proteins, it is clear that this is an issue that needs to be addressed.
Alternative proteomic approaches exist for identifying proteins that interact or function cooperatively. Examples are the yeast two-hybrid system 38 or the tandem affinity purification (TAP) system 39. However, to the present author's knowledge, these have not yet been applied to the tubercle bacilli on any significant scale. Protein/protein interactions can also be predicted in silico using recently developed algorithms, such as those for phylogenetic profiling, in which the distribution of given proteins in target species is examined, or by comparing target bacterial sequences with those of multidomain eukaryotic proteins to detect conserved segments that might indicate intervention in a common pathway 40–42. A series of interesting predictions made by these techniques has been reported 43 and is now being validated experimentally.
Gene replacement
Genes can be efficiently inactivated in tubercle bacilli by means of allelic exchange, using haploid or partially diploid hosts, mediated by suicide vectors carrying the defective allele 44 or by conditionally replicating mycobacteriophages 45. Random gene inactivation can be achieved through conventional or “signature-tagged” transposon mutagenesis 45–47. Sequencing the sites of insertion of large numbers of natural (IS6110) or artificial (IS1096-based) transposons provides information about nonessential genes. The signature-tagged mutagenesis method allows disruption and identification of nonessential genes that are not required in vitro but play vital roles in murine TB models 46, 47. Among several thousand signature-tagged mutants tested in two independent screens, the most attenuated strains harboured transposons in genes involved in lipid metabolism and the synthesis and export of phenolphthiocerol dimycocerosate, a characteristic wax confined to pathogenic mycobacteria. Elimination of the dispensable genes leaves a set of >2,000 genes that are probably essential. However, given the relatively large size of the genome of M. tuberculosis and, above all, its long generation time, it is unlikely that the wholesale inactivation of these genes by allelic exchange will be undertaken. Another means of assessing essentiality involves the use of antisense RNA, but this has not yet seen widespread application in M. tuberculosis. An alternative way of identifying essential genes indirectly is to use comparative mycobacterial genomics.
Comparative genomics of the Mycobacterium tuberculosis complex
Comparative genomics is a powerful new tool for exploring microbial evolution and identifying genes that might encode new drug targets or protective antigens. Genomic diversity of the M. tuberculosis complex has been studied by DNA array technology, facilitated by the fact that all members share a >99.95% identity at the DNA level 48. On examination of different BCG strains, a combined total of 18 deletion regions (RD1–RD18) were uncovered by several laboratories 49–51, some of which were specific for given strains. This has led to revision of the genealogy of BCG and to the finding that 120 genes are present in M. tuberculosis H37Rv but absent from BCG Pasteur. This discrepancy may account for the phenotypic differences between the vaccine and the pathogen. Only one region, RD1, is missing from all BCG strains, yet present in the other M. tuberculosis complex members 49, 50, 52. It is possible that loss of RD1 may account for the attenuation of M. bovis originally described by Calmette 53.
Comparative genomics has also uncovered two tandem duplications of 29 and 36 kb (DU1, DU2) in the chromosome of M. bovis BCG Pasteur, indicating that this vaccine strain is partially diploid for 58 genes 54. The combined findings of these comparative genomic analyses indicate that there is substantial genetic diversity between the various BCG strains in use today, which might account for the variability observed in different vaccine trials against TB.
Comparative genomics of the respective members of the M. tuberculosis complex has revealed the existence of a gene gradient. The human tubercle bacillus, M. tuberculosis, has more genes than M. africanum, M. microti and M. bovis, as these species have lost genetic material through deletion events 50, 55. Gene loss occurs at a high frequency within the species M. tuberculosis as the result of homologous recombination events between copies of IS6110 that flank genes in the direct orientation 50, 55, 56. Microarray and Affymetrix chip studies have uncovered an additional group of 45 genes whose presence, and possibly function, is facultative 51, 57. From the combined findings it can be concluded that >200 genes exist that are not essential for growth of M. tuberculosis complex members in the host but may influence the degree of virulence.
Comparative mycobacterial genomics
An even more powerful means of reducing the number of potential new targets within M. tuberculosis to a more tangible level may be found by comparative genomics with the leprosy bacillus, an exceptionally slow-growing obligate intracellular pathogen that displays essentially the same cellular tropism and host range as the tubercle bacillus. At 3.27 Mb, the genome of M. leprae is substantially smaller than that of M. tuberculosis and a mere 49.5% is occupied by the 1,605 protein-coding genes 15. M. leprae appears to have undergone reductive evolution, a process involving extensive downsizing and gene decay. About 27% of the M. leprae genome contains pseudogenes, inactive reading frames that still have functional counterparts in the tubercle bacillus, of which 1,114 are recognisable, while the remaining 23.5% of the genome appears to be noncoding and is probably nonfunctional. Reductive evolution has been documented in obligate intracellular pathogens and endosymbionts, such as Rickettsia and Buchnera spp., respectively 15. Genes are progressively lost as their functions are no longer required in highly specialised niches and this probably accounts for the exceptionally long generation time of the leprosy bacillus. In summary, assuming that all mycobacteria are descended from a common ancestor, M. leprae has probably lost >2,000 genes during its evolution and the minimal gene set required by a pathogenic mycobacterium may have been defined naturally 15.
When pairwise comparisons of the gene and protein sets of the leprosy and tubercle bacilli 12, 15, 16, 58 were performed, 1,433 proteins were found to be common to both pathogens. After removal of proteins that are shared with all other prokaryotes (except Actinomycetes) and eukaryotes the sample contains only 333 proteins. Since these pathogenic mycobacteria occupy similar niches in the human body, where they encounter the same physiological stresses and immune responses, it is conceivable that the products of some of these genes may affect highly specialised functions that could be essential for intracellular growth of mycobacteria. If this was the case, the corresponding proteins or enzymes might represent novel drug targets. The 333 candidates identified by comparative mycobacterial genomics can be subdivided into those proteins that are confined to the genus Mycobacterium (there are 219 of these) and a second group of 114 polypeptides that also occur in Streptomyces or Corynebacteria spp., related members of the Actinomycetales kingdom. It is reasonable to assume that the latter proteins confer specific properties on actinomycetes, whereas those that are restricted to mycobacteria may play an even more specialised role.
Target discovery
To further illustrate the usefulness of comparative mycobacterial genomics for identifying potentially important proteins, two precise examples will now be given. Multiple gene duplication events have occurred in M. tuberculosis and when limited divergence has followed, this appears to have resulted in extensive functional redundancy in numerous biochemical pathways 12, 16, 58. Indeed, in many cases it is difficult to predict with certitude which of two duplicated genes affects a specific function. This is true for five proteins (Rv0462, Rv0794c, Rv2855, Rv2713, Rv3303c) that show strong similarity on database searches to various lipoamide dehydrogenase components of the pyruvate dehydrogenase complex, an essential function. During the initial analysis of the M. tuberculosis genome, the two proteins displaying the strongest similarity to this dehydrogenase were termed lpdA (Rv3303c) and lpdB (Rv0794c), but subsequent biochemical studies of the various gene products revealed that authentic lipoamide dehydrogenase was encoded by Rv0462 (now termed lpd) and not by the lpdA or lpdB genes 59. Inspection of the M. leprae genome sequence would have helped focus this work considerably, since only one of these five M. tuberculosis genes has a functional orthologue, with ML2387 corresponding to Rv0462, the true lipoamide dehydrogenase. The remaining four genes are present in pseudogene form in the M. leprae genome 15, 60. This is strong testimony to the power of comparative genomics.
The following is a second example that awaits biochemical confirmation. Preproteins exported by the twin-arginine transport, or Tat, pathway generally bind redox cofactors and fold or oligomerise before crossing the membrane 61, 62. After removal of the signal peptide, many of these proteins function in extracytoplasmic electron transfer chains. The specialised machinery that recognises the twin-arginine motif 63 and translocates the preprotein across the membrane is composed of several different Tat proteins. In E. coli, TatA and TatE are 50% identical and share weak similarity with TatB 62. All three proteins are predicted to be anchored to the cytoplasmic membrane via an N‐terminal hydrophobic α‐helix and to have cytoplasmic amphipathic helices followed by variable regions. The TatC protein is predicted to be an integral membrane protein with six transmembrane segments. M. tuberculosis and M. leprae both contain clearly identifiable tatA, tatB, tatC and tatD genes and must, therefore, produce a functional Tat system.
On examination of the proteome of M. tuberculosis, 11 potential substrates for the Tat export system were recognised on the basis of their signal peptides containing potential twin-arginine motifs at the N‐terminus and the cognate motif, S/TRRXFLK 63 (table 2⇓). During the extensive reductive evolution of the genome of M. leprae only one of the corresponding genes, ML1190, escaped inactivation. It is orthologous to Rv2525c of M. tuberculosis but shows no similarity to other proteins present in current sequence databases. The 240 amino acid-long precursor protein encoded by ML1190/Rv2525c contains five histidine and one cysteine residue that may be important for coordinating divalent metal ions. The conservation of this coding sequence by M. leprae, in the face of massive gene loss, is a strong indication that it must play an important biological role. Given the many parallels with Tat systems elsewhere, it is likely to be in electron transport. These indirect arguments suggest on the one hand that, if this function were essential, the ML1190/ Rv2525c gene product might represent a novel drug target or, on the other, since it is likely to be located extracellularly, it may be an important sentinel protein antigen.
Possible twin arginine secreted proteins
Screening drug targets
If the corresponding protein has an assayable function, kinase activity for example, it can be used as the basis of an in vitro screen to identify inhibitors of the enriched or purified enzyme. The advantage of this approach is that it can generally be automated or converted to high-throughput format to facilitate screening of large or complex libraries of synthetic compounds or natural products. However, whole organism screens, involving recombinant M. tuberculosis strains with reporter activity, such as luciferase or green fluorescent protein 64, 65, are often considered preferable as they avoid drug permeability problems. Once an active pharmacophore has been uncovered, numerous analogues can be synthesised or identified in combinatorial libraries to isolate more active derivatives. Their potency can also be evaluated using reporter assays or biochemical techniques, such as transcriptome or proteome analysis. In this way, genes that are coregulated can be uncovered whose products may also serve as potential drug targets in turn, as they often act concertedly in the same metabolic process. Identification of the target enables large amounts of the corresponding protein to be produced by genetic engineering for further studies. Knowledge of the three-dimensional structure of known or potential drug targets is also highly desirable for drug development purposes and can be obtained through structural biology.
Structural genomics
Structural genomics is an emerging scientific discipline that exploits high-throughput cloning technology to generate systematically large numbers of expression constructs corresponding to most, or all, of the genes present in a microbial genome (table 1⇑). These constructs are used to produce large amounts of tagged protein that can be purified in a single step by powerful affinity chromatography methods, thereby facilitating downstream structural analysis by X‐ray crystallography, nuclear magnetic resonance (NMR) spectroscopy or high-resolution cryo-electron microscopy. Current experience predicts a 60% success rate at the expression stage, with ∼20% of purified proteins subsequently giving diffraction grade crystals. Structural genomics is a promising new approach to drug and drug-target discovery as it readily interfaces with structure-based drug design 66. In the case of larger genomes, structural genomics generally concentrates on large protein families to maximise the yield, whereas in drug-target discovery programmes the approach is usually hypothesis-driven.
Diagnostics
The diagnosis of both active disease, which relies heavily on clinical expertise and the detection of acid-fast bacilli in sputum smears, and latent TB will also benefit from comparative genomics. Latent infection is often diagnosed by monitoring the extent of delayed type hypersensitivity reactions following intradermal injection of tuberculin, an ill-defined mixture of antigens. Tuberculin reactivity is of limited value in communities where BCG vaccination is practised and its interpretation may also be confounded by infections involving other mycobacteria. The identification of 120 genes in the tubercle bacillus 49, 50, which are absent from BCG, allows a move towards the development of a more specific test that can distinguish between infection and immunisation. Microarrays and proteomics will also find wide application in monitoring biodiversity within the M. tuberculosis complex and help to confirm the presence or absence of candidate diagnostic antigens.
Antigen discovery and vaccine strategies
Knowledge of the subcellular location of proteins is particularly valuable for the design of new TB vaccines, both preventive and therapeutic, since it is widely believed that surface-exposed or secreted proteins correspond to those antigenic components that are first encountered by the immune system during infection 67. Figure 1⇓ presents a scheme whereby such proteins can be found. The system employs bioinformatics to identify proteins that localise to the cell envelope. These include transmembrane proteins with hydrophobic domains and lipoproteins with N‐terminal cysteine residues that are modified by addition of lipid groups. Proteins that are secreted via the general secretory pathway 68 are readily identifiable by their characteristic signal peptides as are those metallo-enzymes that are secreted by the TAT system 61–63. Other proteins that lack signal peptides and are secreted from mycobacteria in a Sec-independent manner include those belonging to the early secreted antigenic target (ESAT)‐6 family 12, 58. ESAT‐6 is a potent T‐cell antigen that induces strong T‐helper 1‐type responses 69 and has been extensively studied as a potential diagnostic reagent for infection 70, since its gene is missing from BCG 50, 51, 53, 71, and as a component of a subunit vaccine 72. In addition to highly purified proteins that are envisaged for use as subunit vaccine components, other strategies are being pursued. These include nucleic acid vaccines 73, 74, the construction of rationally attenuated mutants of M. tuberculosis, and improved BCG strains 75. All of these approaches have been boosted by the availability of the genome sequence 12.

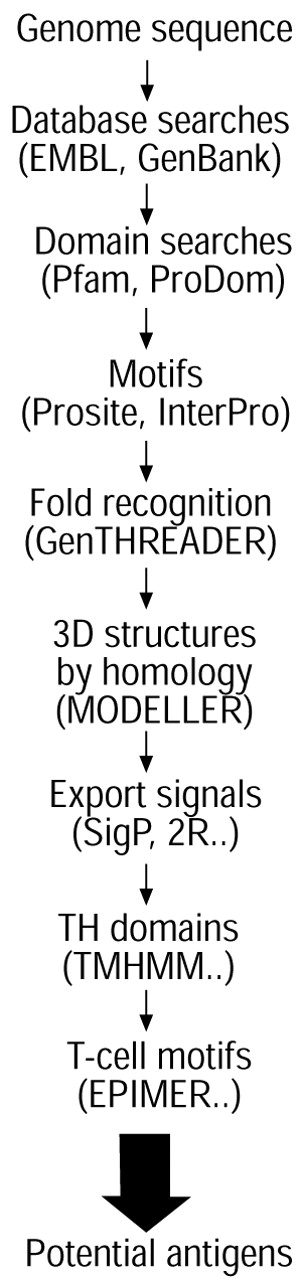{kind=link}
A scheme to identify potential antigenic proteins using bioinformatic tools to analyse genome data. The databases or programs used are shown in brackets. See main text for further details. 3D: three dimensional.
Appraisal of immunogenicity in silico and in vivo
Comparative in silico analysis of the proteome of Mycobacterium tuberculosis has reduced the number of potential subunit vaccine candidates from ∼4,000 proteins to <40 potentially exported polypeptides, which are present in both the leprosy and tubercle bacilli. Several of these belong to protein families of conserved sequence that are invariably larger in Mycobacterium tuberculosis than in Mycobacterium leprae. The comparative genomic analysis has highlighted the potential importance of the esterase activator‐6 proteins 12, 58, together with their likely secretion machinery, and initial immunological characterisation has yielded encouraging results 76. To establish which of these protein candidates contain potential T‐cell epitopes, powerful new algorithms such as EpiMer 77, can be used to further refine the selection. The availability of the genome sequences of both mice and humans 10, 11 will allow potentially crossreactive epitopes or antigens to be identified in silico, thereby limiting possible complications. Ultimately, the success of the approach will require testing the candidates retained in cellular and animal models for tuberculosis, and establishing the appropriate correlates of protection.
Acknowledgments
The author would like to give special thanks to the mycobacterial genomics groups at the Institut Pasteur and the Sanger Centre. Parts of this work were supported by the Wellcome Trust, the Institut Pasteur, the European Community (QLK2-CT-1999-01093, QLRT-2001-02018), and the Association Française Raoul Follereau.
- Received January 28, 2002.
- Accepted March 13, 2002.
- drug targets
- functional genomics
- genomics
- leprosy
- tuberculosis
- © ERS Journals Ltd