



Cytokines and the lung
- G.B. Toews
- Division of Pulmonary and Critical Care Medicine, University of Michigan Hospital, Ann Arbor, MI, USA
- G.B. Toews, Division of Pulmonary and Critical Care Medicine, University of Michigan Hospital, Ann Arbor, MI, USA. Fax: 1 7347644556
Abstract
Cytokines are signal molecules that induce movement, differentiation, growth and death of many cell types. Cytokines generate these effects through interactions with receptors, which relay a signal into the cell triggering a response.
Cytokine-receptor interactions are promiscuous; a combining site of any receptor can bind many ligands. Promiscuity allows for the generation of agonists, alternative ligands that activate a receptor in a way similar to the normal ligands and antagonists, ligands that bind to a receptor, but neutralize the effects of an agonist. Cytokine-receptor interactions induce many diverse (pleiotropic) effects. Cytokine-receptor interactions are redundant; several cytokines can perform the same function.
Mammalian hosts use cytokines to maintain homeostasis and to provide signals crucial to host responses to invading microbes and other injurious agents. Cytokines are the molecular messages, which: 1) initiate and amplify inflammatory and immune responses by recruiting and activating cells; 2) regulate the activation and differentiation of T‐ and B‐lymphocytes, whose functions are crucial to specific cell-mediated immunity; and 3) initiate and regulate local repair processes critical to the resolution of inflammatory responses.
Further studies of cytokines and their receptors should provide a framework for therapeutic interventions in patients with dysregulated inflammatory responses.
A review of cytokines and the lung is a daunting task. The scope of cytokine biology is immense since these molecules have wide-ranging biological effects. More than 190,000 citations that index cytokines have been published in the last decade and >1,800 cytokine manuscripts were published in the past month. A definition of “cytokines” is not simple; most textbooks and reviews do not provide such a definition. For the purpose of this review, cytokines will be defined as a diverse group of protein signal molecules that are produced by a wide variety of cells. Cytokines influence or activate adjacent cell movement, differentiation, growth and death. Cytokines generate these effects through interactions with receptors already present in low density on the cell surface or through upregulation of new receptors. Cytokine receptors relay a signal into the cell triggering a response. Regulation of receptor expression is as important as regulation of cytokine production if pulmonary homeostasis is to be maintained. Networks of cytokines act cooperatively to control normal physiological activity and disease-related threats to homeostasis. Furthermore, the composition of the cytokine milieu changes as disease processes progress and during resolution, remodelling and healing responses.
Characteristics of cytokine-receptor interactions
Promiscuity
The binding of cytokine ligands to cytokine receptors is promiscuous. A binding site of a receptor can bind many ligands and a ligand can bind several receptors. Molecular specificity hinges on the array of noncovalent bonds formed at the points of contact between the ligand and receptor. Sensitivity of ligand binding to ligand concentration allows the quantitation of binding affinity, the energy of binding between the receptor and the ligand. If the noncovalent forces of attraction between receptor and ligand are great, dissociation is unlikely even at a low concentration of ligand and the binding site of the receptor will be occupied by the ligand. Conversely, low-affinity interactions lead to high rates of ligand dissociation. A high concentration of ligand molecule will be necessary to maintain occupancy. While this degeneracy is disappointing at first glance, the intrinsic degeneracy of cytokine-receptor interactions has important biological consequences. Biological recognition and regulation occurs when molecules and receptors fit and bind for a moment in time. Ligand-receptor interactions, like an effective handshake, are reversible. This ligand-receptor plasticity underlies the bulk of pharmacological approaches to this system of biological regulators. Agonists are alternative ligands that can activate a receptor in a way similar to that of the natural ligand. Antagonists are ligands that bind physically to the receptor, but rather than activating the receptor, neutralize the effects of an agonist. An antagonist can function as a blocking or competing molecule.
Pleiotropia
A single cytokine molecule can produce many diverse effects. Pleiotropia does not refer to the alternative reaction states of a molecule but to a true diversity of function. Cytokines cause effects by activating genes. Interferon gamma (IFN‐γ) activates >200 different genes 1. Each of the 200 genes is more or less pleiotropic themselves; each gene might express itself differently in different target cells. A single molecule such as IFN‐γ is pleiotropic at the most basic causal level.
Redundancy
Redundancy describes situations in which several agents perform the same action. Although the agents are not identical, they are redundant to the degree to which they may replace one another functionally. Chemokines are a particularly redundant cytokine system 2. Redundancy is complicated by the fact that different agents can produce some of the same effects as well as different ones. Tumour necrosis factor alpha (TNF‐α) and IFN‐γ partially overlap in their immune effects; both cytokines are crucial to protective immunity, but each cytokine produces effects that the other does not. Redundancy can be defined as the partially overlapping pleiotropisms of diverse agents.
Polyspeirism
Polyspeirism is a particular type of redundancy. Polyspeirism defines a circumstance in which multiple cytokines are produced in a redundant way by a single cell concomitantly in response to the same stimulus. Polyspeirism is particularly striking for mononuclear phagocytes and endothelial cells exposed to bacterial products. In response to bacterial products, these cells produce virtually all CC chemokines, fractalkine (CX3C) and some CXC chemokines. This characteristic provides a robustness for certain necessary functions such as phagocyte recruitment. Because of the robustness of the response, variations in the quantity of any chemokine(s) or receptor(s) would have bearable consequences for basal trafficking of phagocytes.
Cytokines and the lung
Cytokines, broadly defined, are involved in normal regulation of all physiological processes. The role that cytokines play in the regulation and modulation of immunological and inflammatory processes is striking. The lung is particularly dependent on tightly regulated immunological and inflammatory processes since it is exposed to a large and varied burden of infectious agents as well as a diverse group of noxious gases and particulates during the process of gas exchange. The lung defends itself from these injurious agents by deploying cytokine-regulated host defence mechanisms wherever it interfaces with the external environment 3. This review focuses on the cytokines present at the interface of infectious and inflammatory diseases. Cytokines present in the lung clearly regulate both the initiation and maintenance of immune and inflammatory responses. Cytokines select the type of response generated and the effector mechanisms to be utilized. Cytokines are the intercellular words that provided instructions in cell networks, which provide three crucial functions. First, these cytokine-controlled networks of cells must “classify” the universe into injurious and innocuous agents. Second, the network must “determine” whether to respond or ignore the substance (activation versus tolerance). Third, it must “regulate” the recruited and activated cells 4–7. Cytokines are the molecular signals for communication between cells of the immune system and the systemic mediators or the host response to infection. Cytokines: 1) initiate and amplify inflammation; 2) induce T‐cell independent macrophage activation; 3) regulate dendritic cell maturation and differentiation; 4) regulate T‐cell activation and activation and differentiation; 5) modify connective tissue structures; and 6) regulate blood vessel growth.
Mechanisms of innate immune recognition
The immune system has traditionally been divided into innate and adaptive components 4, 7, 8. Innate immunity is an ancient system of host defence that preceded the development of specific immunity. All multicellular organisms have some form of innate immune defence. The main distinction between the innate and specific immune systems lies in the receptors and mechanisms used for immune recognition. Innate immune recognition is mediated by germ line-encoded receptors. Each receptor has been genetically predetermined to have defined specificities for infectious micro-organisms by natural selection.
Microbial recognition is problematic because of their high mutation rate and their molecular heterogeneity. Eukaryotic organisms must limit the number of genes they commit to microbial recognition. The innate immune response has evolved to recognize a few, highly conserved structures present in large groups of micro-organisms 5, 6. The innate immune system utilizes receptors thought to number in the hundreds to accomplish this task. The receptors recognize molecular patterns rather than particular structures and accordingly have been termed pathogen recognition receptors (PRR) 5–9. The patterns recognized by PRR have been termed pathogen associated molecular patterns (PAMP). Lipopolysaccharides and teichoic acids shared by Gram-negative and Gram-positive bacteria respectively; mannans, conserved components of yeast cell walls; and the unmethylated Poly-cysteine guarine (CpG) motif characteristic of bacterial, but not mammalian deoxyribonucleic acid (DNA), are characteristic PAMPs. While PAMPs are chemically quite distinct, they share certain features. PAMPs are: 1) produced only by microbes and not eukaryotic hosts; 2) essential for pathogenecity or survival of the microbe; 3) invariant structures shared by classes of pathogens 6–9.
The receptors of the innate immune system differ from antigen-specific receptors in several important ways. PRR's are expressed on many effector cells of the innate immune system. All professional antigen-presenting cells, macrophages, dendritic cells, bear PRR's. Second, expression of PRR's is not clonal; all such receptors displayed by cells of a given type have identical specificities. Third, following recognition of PAMP's, PRR's trigger effector cells to perform their effector functions immediately rather than after cellular proliferation 8.
PRR's belong to several families of proteins 10–35. Functionally, PRR's can be divided into three classes: secreted, endocytic, and signalling receptors. The best characterized secreted PRR is the mannan-binding lectin, a member of the calcium-dependent lectin family that binds to microbial carbohydrates to initiate the lectin pathways of complement activation 23, 24.
Secreted PRR's function as opsonins by binding to microbial cell walls and identifying microbes for recognition by the complement system and phagocytes. The macrophage-mannose receptor, also a member of the calcium-dependent lectin family, is an endocytic PRR 11, 24. It specifically recognizes carbohydrates with large numbers of mannoses that are characteristic of micro-organisms and mediates their phagocytosis by macrophages. The macrophage-scavenger receptor, another endocytic PRR, binds to bacterial cell walls and is an essential part of the clearance of bacteria from the circulation 20, 21, 25, 26. Signaling PRR's recognize PAMP and activate signal transduction pathways that induce the expression of a variety of immune response genes including inflammatory cytokines and costimulatory molecules. Toll-like receptors (TLR) are signalling PRR's 27, 28. Optimal signalling efficiency of TLR family members depends on association of secreted proteins MD‐1 or MD‐2 29.
The mammalian toll family contains at least 10 family members. Different classes of pathogens are recognized by different, specialized members of the toll family. TLR4 appears to be responsible for the detection of Gram-negative bacteria. Recognition of lipopolysaccharide (LPS), a PAMP that represents a molecular signature of Gram-negative bacteria, mediates this detection process 30–32. TLR2 is involved in recognition of Gram-positive bacteria through recognition of lipoteichoic acid and peptidoglycan 33. TLR6 cooperates with TLR2 in detecting a subset of bacterial peptidoglycan 34. TLR9 detects bacterial DNA sequences containing unmethylated cytosine-guanosine dinucleotides (CpGs) 35. The specificity of the other six mammalian TLRs is the subject of intense investigation. Other members of the mammalian TLR family will most likely be specific for PAMP's characteristic of other classes of pathogens such as fungi (mannan, glucan and mycobacteria (lipoarabinomannan, muramyldipeptide)).
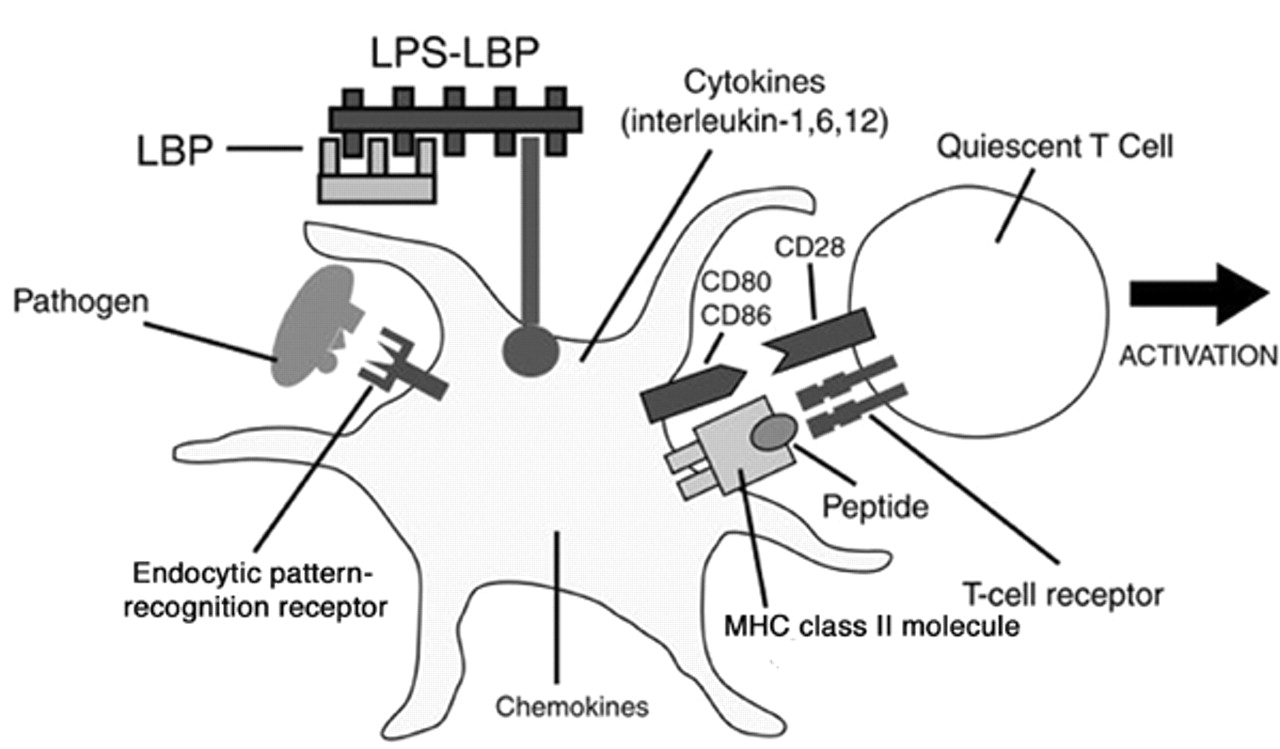
Activation of TLRs initiate signal transduction pathways that initiate and amplify inflammatory responses in the lung and modulate adaptive immune responses. The recognition and signalling pathways involved in the recognition of LPS have been most extensively studied. LPS initially interacts with a serum protein LPS binding protein, which transfers LPS to CD14, a receptor on macrophages and B‐cells. MD‐2, a cell surface protein, is required for TLR4 mediated recognition of LPS. Accordingly, the LPS recognition complex most likely consists of three molecular components; CD14, TLR4, and MD‐2 14, 29, 36. The binding of LPS to CD14 presumably leads to the association of CD14 with the TLR4‐MD‐2 complex. Assembly of this complex may induce the dimerization of TLR4. TLR4 activation results in recruitment of the adapter protein MyD88, which is associated with the serine-threonine protein kinase interleukin‐1 receptor-associated kinase (IRAK). This association leads to IRAK phosphorylation and association with the tumor necrosis factor-associated factor 6 (TRAF‐6) adapter protein. Oligomerization of TRAF‐6 then activates a member of the mitogen-associated protein kinase, kinase, kinase (MAP3K) family which directly or indirectly leads to the activation of IκB kinase 1 (IKK1) and IκB kinase 2 (IKK2). These kinases target IκB for degradation and release nuclear factor (NF)-κB, which moves into the nucleus and induces transcriptional activation of a wide-variety of inflammatory- and immune-response genes. Molecules induced by LPS-TLR4 interactions include: 1) signals that generate inflammatory responses including TNF‐α, interleukin (IL)‐1, IL‐6, interferon (IFN) α/β and chemokines; 2) signals that function as co-stimulators of T‐cell activation, CD80 and CD86; and 3) signals that regulate the differentiation of lymphocytes including IL‐4, IL‐5, IL-10, IL-12, transforming growth factor (TGF)‐β and IFN‐γ 8.
Inflammatory responses
The generation of inflammation in the lower respiratory tract involves the co-ordinated expression of both pro- and anti-inflammatory cytokines (fig. 1⇓). Following the recognition of microbial products, TLR-mediated signalling results in the production of TNF‐α and IL‐1β 37–42. Neutrophil recruitment is dependent on the orchestrated generation of these two early-response cytokines. TNF‐α and IL‐1β stimulate expression of adhesion molecules on vascular endothelial cells 43, 44. P‐ and E‐selectins expressed on endothelial cells interact with L‐selectin on neutrophils leading to neutrophil rolling. Intercellular adhesion molecule (ICAM)‐1 expression is also induced on the surface of the endothelium. Firm adhesion results following interactions between neutrophils and ICAM 43, 44.
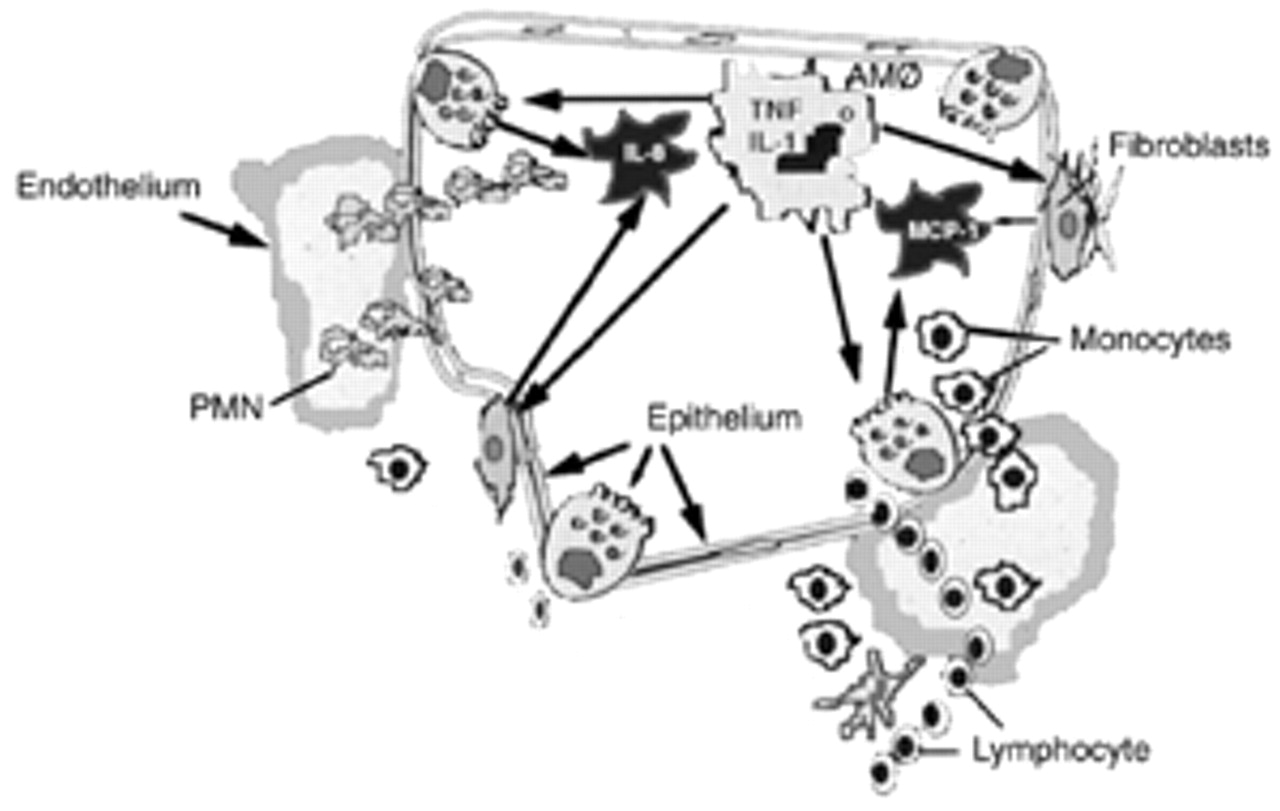
Development of an inflammatory response. Innate immune cells such as alveolar macrophages and monocytes recognize microbial products and secrete chemotaxins which recruit polymorphonuclear neutrophils (PMN's) to the airspace. Innate immune cells also secrete early response molecules such as interleukin (IL)‐1 and tumour necrosis factor (TNF), which activate alveolar epithelial cells and fibroblasts to produce chemokines. The development of an inflammatory response requires the induction of a coordinated network of cytokines, which involves cells throughout the alveolo-capillary wall. AM: alveolar macrophages; MCP: monocyte chemoattractant protein.
CXC chemokines are also rapidly produced following microbial stimuli. IL‐8, growth regulated oncogene (GRO) and epithelial-derived neutrophil attractant (ENA)-78 are produced by mononuclear phagocytes, neutrophils and endothelial cells in response to LPS, TNF‐α and IL‐1β 45–52. IL‐8, GRO and ENA-78 are all major polymorphonuclear neutrophil (PMN) chemoattractants. Expression of these CXC chemokines in the lung is stimulus specific. Thus, a network of cytokines rapidly induces PMN accumulation within the lung following recognition of microbial products 50.
Resident alveolar macrophages and recruited monocytes play a major role in amplifying the inflammatory response in the lower respiratory tract. Macrophages amplify the inflammatory response by stimulating cytokine production by cells that do not respond directly to bacterial products. Pulmonary fibroblasts, various pulmonary epithelial cells and pleural mesothelial cells all produce IL‐8 in response to specific host-derived signals such as TNF‐α or IL‐1β 48–50. All nucleated cells possess a functional receptor for IL‐1 and TNF‐α suggesting the importance of IL‐1 and TNF‐α as key cytokines in the augmentation of inflammatory responses. TNF‐α and IL‐1 induce all major cellular components of the alveolar-capillary membrane and/or airway of the lung to participate in the inflammatory response. Directed migration of leukocytes into the lung is dependent on the co-ordinated, sequential expression of chemokines.
T‐cell independent macrophage activation
T‐cell independent macrophage activation is one of the earliest events in the innate immune response. Cell-cell communication between macrophages and natural killer (NK) cells is required during this activation process 53. Following microbial recognition and TLR-mediated signalling, macrophages release IL-12 and TNF‐α. These cytokines, in synergy, induce IFN‐γ production by NK cells. IFN‐γ is responsible for priming macrophages for microbicidal activity 54–57. Both positive and negative feedback mechanisms regulate production of IL-12. IFN‐γ, primarily an NK cell product, is the most potent up-regulator of IL-12 production. IL-10, a product of macrophages and other cell types, is a potent inhibitor of IL-12 production 58. IL-10 expression is delayed compared to that of IL-12 in vivo. This difference in kinetics is crucial to the role of IL-10 as a defence against excessive production of IL-12 and its pathological consequences.
Both LPS from Gram-negative bacteria and lipoteichoic acid from Gram-positive bacteria induce IL-12 via TLR-dependent mechanisms 59, 60. IL-12 production is also induced by bacterial DNA; CpG repeats are crucial for this induction 61, 62. Dendritic cells can also be stimulated to produce IL-12 via TLR-mediated signalling pathways 63.
IL-12 is important to innate immune defence mechanisms in the setting of murine models of bacterial pneumonia 64. Overexpression of IL-12 messenger ribonucleic acid (mRNA) and protein within the lung during bacterial pneumonia results in a 35% long-term survival in a model in which no animals that receive control adenovirus survive. In vivo inhibition of IL-12 bioactivity results in significant impairment in bacterial clearance and results in increased mortality. The mechanism by which compartmentalized overexpression of IL-12 improves survival is not completely understood. Treatment with anti-IFN‐γ‐antibodies or a soluble TNF receptor-Ig construct partially and completely attenuates survival benefits observed in animals receiving adenoviral vector-delivered IL-12. These studies indicate that IL-12 is an integral cytokine in innate immune responses against Gram-negative bacterial pathogens.
The delayed expression of IL-10 compared to that of IL-12 in vivo make it a likely, effective down-regulator of the IL-12 response 65. Blockade of IL-10 adversely affects the outcome of intravenous LPS challenges in mice 66. IL-10 -/- mice infected with Toxoplasma overproduce IL-12, which results in a lethal syndrome 67. In aggregate, these studies suggest that IL-10 is a critical defence against excessive production of IL-12 and its pathological consequences.
Dendritic cell maturation and differentiation
Antigen presenting cells (APC) provide an essential link between innate and adaptive immunity. Dendritic cells (DC) are the most capable initiators of adaptive immune responses 68, 69. DC are localized at epithelial borders throughout the mammalian host where they recognize pathogens and/or micro-environmental tissue damage and signal the presence of “danger” to cells of adaptive immunity 7, 70, 71. DC capture antigen, migrate to draining lymphoid organs and, after a process of maturation, select antigen-specific lymphocytes to which they present processed antigen, thereby initiating adaptive immune responses 68, 69.
DC are believed to be a complex system of cells encompassing multiple subsets with potentially distinct biological functions 69, 72–81. DC comprise three distinct populations, including two within the myeloid lineage, Langerhans cells and interstitial DC and one within the lymphoid lineage. Within these pathways, DC at different maturational stages may differ in phenotype, function and localization. Three stages of maturation have been delineated including precursor DC found in blood and lymphatics, tissue-residing immature DC, and mature DC present within secondary lymphoid organs 72, 82–89.
Dendritic cell development from blood precursors
The study of DC biology has been impeded by the rarity of DC in vivo and difficulties in purifying DC. Considerable progress has been made in understanding DC maturation by studying human DC in vitro.
DC progenitors represent a small fraction of CD34+ haematopoietic progenitor cells in bone marrow or peripheral blood. Granulocyte macrophage colony stimulating factor (GM-CSF) and TNF‐α stimulate growth and differentiation of DC progenitors into DC precursors. This process can be enhanced and/or modulated by multiple cytokines including c‐KIT ligand, Flt‐3‐ligand, IL‐3, TGFβ, IL‐4 and IL-13. TNF‐α, however, appears to be a critical factor in DC development 73, 90. Myeloid DC are closely related to monocytes. Monocytes generate myeloid DC when cultured with GM-CSF and IL‐4 (IL-13). Conversely, immature myeloid DC differentiate to macrophage phenotypes when cultured with GM-CSF 91–95. The choice of whether a monocyte becomes a DC or a macrophage may in part be influenced in vivo by the endothelium. Monocytes that transmigrate the endothelium in the ablumenal-to-lumenal direction (during entry into lymphatics) become DC's; those that remain in the tissues become macrophages 96.
Dendritic cell migration and maturation
Chemokines and their receptors play a major role in directing the right cells to the right places (fig. 2⇓). Immature DC's as well as monocytes, which represent immediate DC precursors, express various receptors for inflammatory chemokines such as CCR1 (receptor for regulated on activation, normal T‐cell expressed and secreted (RANTES), CCR2 (receptor shared by monocyte chemoattractant protein (MCP)‐‐1‐MCP4), CCR3 (receptor for eotaxin), CCR5 (receptor for monocyte inflammatory protein (MIP)‐1α and ‐β and RANTES), and CCR6 (receptor for MIP‐3α) 97–101. As a result of these receptor-ligand interactions, monocytes and immature DC are rapidly recruited to organs undergoing inflammatory responses. LPS or TNF‐α stimulate DC to produce large amounts of inflammatory chemokines, which contribute to the further recruitment of immature DC's. After arrival at sites of inflammation, DC capture antigens and are stimulated to mature by inflammatory cytokines such as TNF‐α or IL‐1 or by bacterial or viral products such as LPS, CpG or double-stranded ribonucleic acid (RNA) 102.
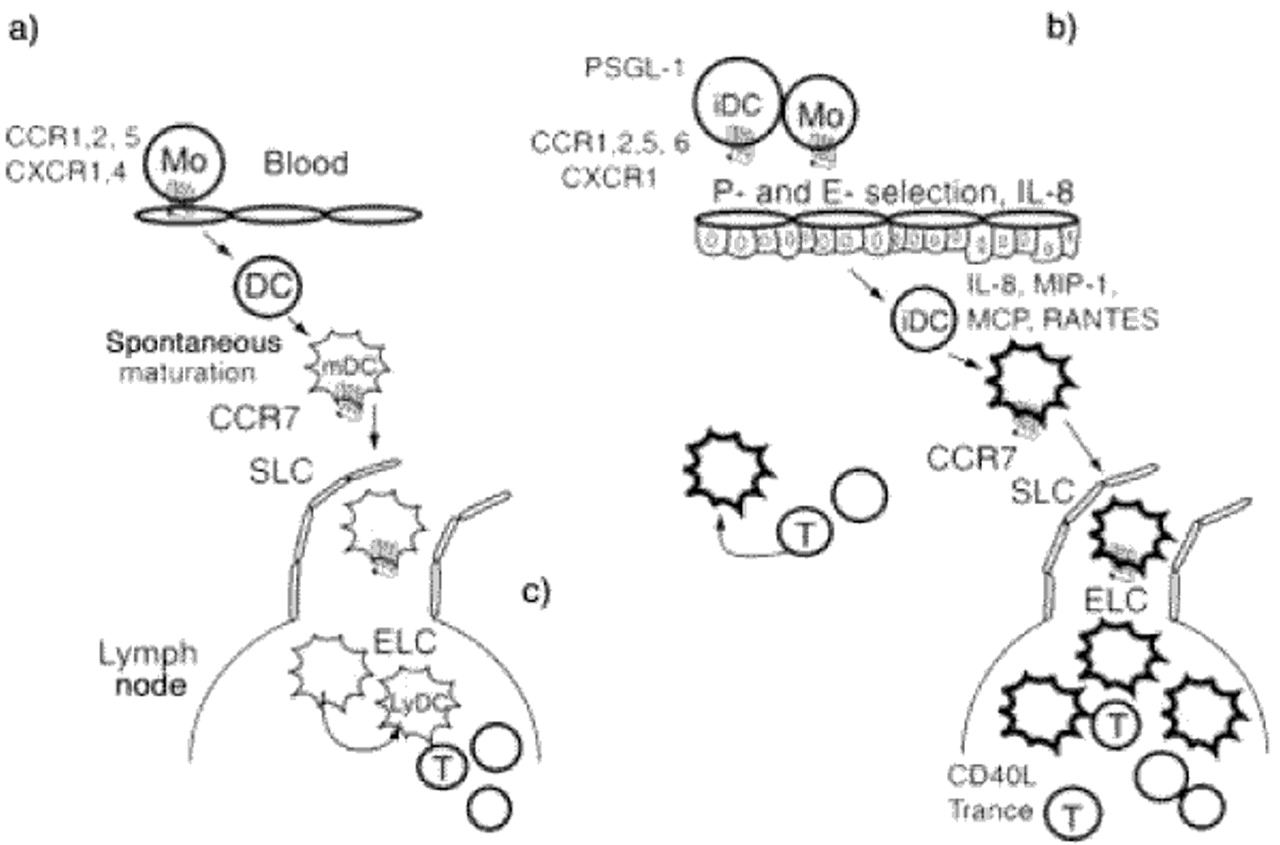
Migration and activation of dendritic cells (DC) controls the induction of immune responses and chronic inflammation. a) tolerance, b) priming, c) chronic inflammation. Immature DC are attracted by inflammatory chemokines into sites of inflammation. Microbial products induce DC maturation and activation. Chemokines induce DC migration to draining lymph nodes. In the lymph nodes, activated T‐cells further enhance DC activation and survival via CD40L and TRANCE. The presence of many highly-activated DC induce a productive T‐cell response within the node. Mature DC that fail to migrate to lymph nodes and remain in peripheral organs may serve as initiation sites for chronic inflammatory responses. Mature DC produce chemokines in peripheral organs to attract maturing DC as well as recently activated T‐cells. In aggregate, these cells maintain a chronic inflammatory reaction. RANTES: regulated on activation, normal T‐cell expressed and secreted; TRANCE: tumour necrosis factor-related activation-induced cytokine; CCR: chemokine receptor; MO: monocyte; MDC: macrophage-derived chemokine; SLC: secondary lymphoid chemokine; ELC: EBII-ligand chemokine; MIP‐1: macrophage inflammatory protein; MCP: monocyte chemoattractant protein.
The maturation process leads to an upregulation of co-stimulatory molecules (fig. 3⇓). T‐cells require at least two signals to become activated; one is a complex of a peptide and a major histocompatibility complex (MHC) molecule, and the other is a co-stimulatory signal mediated by molecules such as CD80 and CD86 on the surface of APC. It is only when the APC expresses both antigen and CD80 or CD86 molecules that T‐cells can be activated. Recognition of an antigen in the absence of DC80 or CD86 molecules leads to permanent inactivation or apoptosis of T‐cells 103, 104.

Receptors and cytokines involved in innate immune control of adaptive immune responses. Endocytic pattern recognition receptors such as the mannose receptor, bind to components of microbes and mediate the phagocytosis of pathogens by antigen presenting cells (APC). Ingested micro-organisms are processed to general antigenic peptides, which complex with major histocompatibility complex (MHC) class II molecules on the surface of APC. MHC-microbial peptide complexes are recognized by T‐cell receptors. Pathogen associated molecular patterns (PAMP's) recognized by Toll-like receptors activate APC to express cytokines, chemokines and co-stimulatory molecules. Pathogen recognition receptors (PRR's) play crucial roles in the generation of both peptide-MHC molecule complexes and the expression of co-stimulatory molecules required for activation of T‐cells. LPS: lipopolysaccharide; LBP: lipopolysaccharide binding protein.
The expression of CD80 and CD86 molecules on the surface of APC is controlled by innate immune system signals 27. TLR's induce these molecules to appear on APC when they recognized their cognate PAMP's. Since PAMP's occur only on pathogens, TLR's induce CD80 and CD86 molecules only in the presence of infection. Accordingly, a T‐cell receives both of the signals required for activation only if its receptor binds to the peptide that was derived from the pathogen that induced the expression of CD80 or CD86 molecules through its PAMP (i.e. LPS). This mechanism insures that, normally, only pathogen-specific T‐cells are activated. Innate immune recognition, thus, controls all major aspects of the adaptive immune response through the recognition of infectious microbes and the induction of signals required for the triggering of specific immunity.
The maturation process also leads to migration to secondary lymphoid organs. As a consequence of chemokine production, maturing DC's down-regulate CCR1, CCR5 and CCR6 100, 101. Conversely, receptors for constitutive chemokines CCR4, CCR7 and CXCR4 are upregulated in maturing DC. CCR7 is most likely of particular importance to the movement of mature DC into lymphatics since the ligand secondary lymphoid tissue chemokine (SLC) is produced by lymphatic endothelial cells 105. From the lymphatics, DC's drain to the lymph nodes where final positioning within the T‐cell area may be controlled by other CCR7 ligands including ELC, produced by resident mature DC and MIP-3β 106.
DC are important sentinels of the immune system in the lung 107 (fig. 4⇓). Airway and alveolar epithelial cells most likely participate in the differentiation of DC in the lung by secreting cytokines as a direct response to pathogens and secondly, by acting as paracrine cells, which amplify ongoing airway and parenchymal inflammation through the production of cytokines in response to inflammatory mediators. Micro-environmental airway or alveolar tissue injury might also initiate the activation of pulmonary DC. The precise signals that injured cells might deliver are uncertain but could include cytokines, oxygen radicals or altered glycosylated cell-surface molecules. Pulmonary epithelial cells and DC most likely participate in a differentiation network within the innate immune system 107–109.
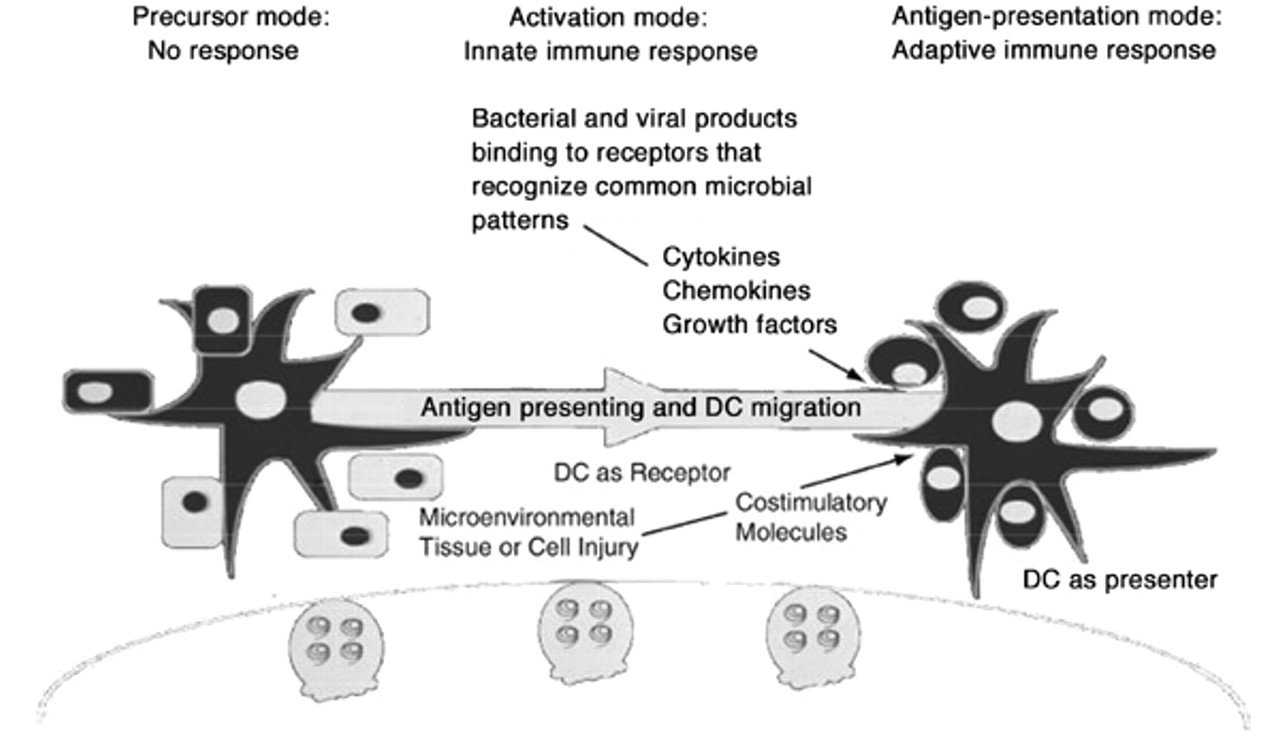
The immunological role of lung dendritic cells (LDC). Pulmonary parenchymal and mesenchymal cells present in the airway and lower respiratory tract participate in the differentiation of dendritic cells by releasing cytokines, chemokines and growth factors following exposure to microbial products or as a result of micro-environmental cell injury. These cytokines, chemokines and growth factors lead to: 1) migration of DC from blood to lung; 2) maturation/activation of DC within the lung; and 3) migration of DC to the node. Dendritic cells are sentinels of the immune system in the lung.
T‐cell differentiation
Naïve T‐lymphocytes differentiate into distinct effector cells depending on the cytokine milieu they encounter during activation (fig. 5⇓). Two subsets of CD4 and CD8 T lymphocytes have been defined on the basis of their lymphokine secretion patterns. T1 cells generate IL‐2, IFN‐γ, TNF‐α and GM-CSF; T1 cells mediate delayed-type hypersensitivity responses. T2 cells produce IL‐4, IL‐5 and IL-10; T2 cells are responsible for B‐lymphocyte maturation and immunoglobulins isotype switching 110, 111.

Regulation of T‐lymphocyte responses. Cells of the innate immune system recognize pathogen associated molecular patterns (PAMP's) and secrete cytokines, which regulate T‐subset differentiation. The cytokine milieu in which naive T‐cell differentiation occurs determines the eventual cytokine profile and effector function of T‐lymphocytes. The innate immune system and its cytokines are crucial determinants of the type of effector cells generated during adaptive immune responses following antigen challenge. IL: interleukin; GM-CSF: granulocyte macrophage colony stimulating factor; PA: plasminogen activator; PAI: plasminogen activator inhibitor; TGF: transforming growth factor; PDGF: platelet derived growth factor; IGF: insulin-like growth factor; KGF: keratinocyte growth factor; HGF: hepatocyte growth factor; PGE2: prostaglandin E‐2.
IL-12 is the single most important factor required for the efficient differentiation of T1 cells 112–114. IL-12 is induced by bacteria, fungi, viruses or their products by a T‐independent pathway and by a T‐cell-dependent pathway mediated through CD40-CD40 ligand (CD40L) interactions 59, 63, 115, 116. IL-12 and IFN‐γ induced by IL-12, create a micro-environment during the early inflammatory phase of an infection in which antigen-specific CD4 and CD8 T‐cells are preferentially induced to differentiate into T1 cells that produce even higher levels of IFN‐γ 117–119.
IL-12 is a 75 kDa heterodimer produced by macrophages and DC 120, 121. IL-12 directs T1 development from antigen-stimulated naïve T‐cells and activates signal transducers and activators of transcription (STAT)3 and STAT4 in T1 cells. Gene deletion of IL-12 or STAT4 markedly reduces T1 responses demonstrating that IL-12 signalling through this pathway is required in vivo 122–125. Functional receptors for IL-12 appear to be restricted to recently activated, uncommitted cells and to T1 cells. These receptors are lost during the development of T2 cells 124, 126. T1 development is also dependent on IFN‐γ. The effects of IFN‐γ may be mediated via its action on the macrophage to upregulate IL-12 production or by direct effects on the T‐cell. The molecular basis of IL-12 responsiveness is in part dependent upon the expression of IL-12R. IL‐4 down-regulates expression of IL-12Rβ2; IFN‐γ upregulates expression of IL-12Rβ2 124, 126, 127. IL-18 is also importantly involved in T1 development. This cytokine promotes proliferation in IFN‐γ production by T1 clones and lines and NK cells. IL-18 belongs to the IL‐1 family. IL-18 does not drive T1 development, but potentiates IL-12-induced T1 development. IL-18 does not activate STAT4 in Th1 cells. IL-18 signals through the IRAK pathway 128.
Induction of TNF‐α appears to be a critical early step in the afferent phase of T1 cell-mediated immunity against certain microbes. A single dose of anti-TNF‐α antibodies at the onset of a moderately virulent Cryptococcus neoformans infection results in a four-log increase in lung colony forming units (CFU's) and a five-log increase in brain CFU 5 weeks later. Delaying the induction of TNF‐α alters the usual, protective T1 response induced by C. neoformans to a nonprotective T1 response. These data suggests that TNF‐α induced activation of macrophages or DC is required for effective production of IL-12 by these cells 129, 130.
Chemokine signalling also plays a role in T1/T2 immune response polarization within the lung. In mice lacking CCR2, the primary receptor for MCP‐1, intratracheal inoculation with C. neoformans results in delayed macrophage and CD8 T‐cell recruitment as well as a lack of cryptococcal clearance. CCR2-deficient mice exhibit significant dissemination of C. neoformans to the spleen and brain at 6 weeks postinfection. In contrast to the T1-type response generated by CCR2 expressing mice, CCR2-deficient mice produce a strong T2 response to pulmonary C. neoformans infection. CCR2 -/- mice generate an immune response characterized by chronic pulmonary eosinophilia, crystal deposition in the lungs, pulmonary leukocyte production of IL‐4 and IL‐5, but not IFN‐γ and increased serum IgE. In aggregate, these results demonstrate that expression of CCR2 is required for the development of a T1-type response to C. neoformans infection. Lack of CCR2 results in a switch to a T2-type response 131.
CCR2 deficient mice evidenced defective macrophage and CD8+ T‐cell recruitment to the lung. CD8 depletion during C. neoformans infections results in the production of predominantly T2 cytokines by CD4+ T‐cells 132. Thus, IFN‐γ production by CD8+ T‐cells might be important for the development of T1-type CD4 T‐cell immunity to C. neoformans. Alternatively, CD8 T‐cell produced IFN‐γ may be required to prime macrophages or DC for IL-12 production. Lack of recruitment of appropriate DC to the lung might also explain the CCR2-induced effect.
The development of T2 responses requires IL‐4 during priming of naïve T cells. Cellular sources of this cytokine other than T2 cells are uncertain. Basophils, mast cells, γδ T‐cells and an NK1.1+, CD4+, CD8‐T‐lymphocyte have all been reported to produce IL‐4 133.
Distinct murine DC subsets and their growth factors can direct specific T‐cell subset development. CD8α+ DC elicit T1 responses while CD8α- DC favour T2 responses 85, 134. Repeated injections of either CD8α+ or CD8α-DC yield strongly polarized T1 and T2 responses respectively. CD8α+ DC can be induced to secrete IL-12, which appears essential for their T1 induction 72, 135, 136. The DC molecules that induce T2 responses are unknown, although IL-10 is a candidate molecule. CD8α+ DC are induced to secrete IFN‐γ by IL-12 137. Both DC subsets can efficiently prime antigen specific CD8+ T‐cells and induce cytotoxic T‐lymphocyte activity in vivo 138. It is unknown whether such DC induce distinct cytokine profiles in CD8 T‐cells.
Differential expression of co-stimulatory molecules may also be crucial to this polarization process 139, 140. CD86 is constitutively expressed on DC and macrophages whereas CD80 is not. The outcome of CD80 and CD86 co-stimulation is different. CD86 co-stimulates the production of IL‐4 as well as IL‐2 and IFN‐γ. Thus, after CD86 co-stimulation, an initial source of IL‐4 is available. CD86 co-stimulation provides only a moderate signal for T2 cell differentiation; additional signals are almost surely required to achieve high levels of IL‐4 production.
Multiple pathways have evolved to manipulate the immune response to microbes. Functionally distinct DC subsets exist, but plasticity in DC function exists within these subsets. A system in which DC function was not plastic would not allow the flexibility to evolve differential T‐cell responses during the course of an infectious process. The optimal situation for T‐lymphocyte differentiation would be a circumstance where functionally different DC subsets are geographically segregated and poised to mount a given T‐cell subset response rapidly in response to a given product. However, as the response evolves, the function of these DC may be modified directly, by the pathogen itself, or by cytokines released by neighbouring bone marrow-derived cells, mesenchymal cells or parenchymal cells. The final T‐cell response is accordingly determined by: 1) the microbial product, 2) the receptor on the DC through the which the microbial product signals, 3) the DC subset itself and 4) the local cytokine micro-environment produced by neighbouring bone marrow-derived cells, mesenchymal cells and parenchymal cells.
T‐cell dependent macrophage recruitment and activation
The protective delayed type hypersensitivity response relies on the prompt recruitment of mononuclear cells from the blood in response to chemotaxins produced by both specific T‐lymphocytes and nonspecifically activated cells 141, 142. Mononuclear cell recruitment prevents systemic dissemination of the microbe from the lungs and is also required for effective intrapulmonary microbicidal activity. Chemokines play a crucial role in the recruitment of mononuclear cells during the specific immune response to C. neoformans 143, 144. Intrapulmonary levels of MCP‐1 and MIP‐1α increase following intratracheal inoculation of C. neoformans. MCP‐1 levels are increased during the first 1–2 weeks of infection, whereas MIP‐1α increases 2–3 weeks after infection. MCP‐1 and MIP‐1α are both crucial to the efferent phase of the immune response since administration of neutralizing antibodies to either MCP‐1 or MIP‐1α results in a three-fold increase in fungal burden at 2 weeks postinfection. Neutralization of MCP‐1 abolishes the recruitment of macrophages and reduces CD4+ T‐cell recruitment. Similarly, MIP‐1α neutralization reduces macrophage and neutrophil recruitment significantly. Induction of MIP‐1α production is largely dependent on MCP‐1 production given that MCP‐1 levels were not affected by neutralization of MIP‐1α but neutralization of MCP‐1 significantly decreased MIP‐1α within the lungs of infected mice. Thus, MCP‐1 and MIP‐1α comprise a chemokine network crucial to the maximal recruitment of mononuclear phagocytes during the effector phase of a cell-mediated immune response.
Tissue remodelling and repair
The pulmonary parenchyma and airways must be remodelled and repaired following microbial infection and following injury. Repair and fibrotic processes are crucially dependent on growth factors for fibroblasts and epithelial cells including platelet-derived growth factor (PDGF), transforming growth factor alpha and beta (TGF‐α, TGF‐β), insulin-like growth factor and GM-CSF 145–151 (fig. 6⇓). PDGF induces fibroblast proliferation and collagen production. The wound-healing effects of PDGF are macrophage-dependent in most models. TGF‐α stimulates the closure of wounds induced in cultures of type II alveolar epithelial cell in vitro. TGF‐α most likely plays a role in epithelial cell repair in the lungs following injury 148, 149. TGF‐β has important effects on the turnover of matrix proteins and the proliferation of fibroblasts. TGF‐β activates genes that favor the production on matrix proteins and down-regulates the production of matrix metalloproteinases that digest matrix in the interstitium and alveolar spaces. In aggregate, the effects of TGF‐β shift the balance in favour of matrix accumulation 151. GM-CSF has a protective effect on repair processes following bleomycin-induced injury 152, 153. The protective effect is mediated in part by GM-CSF-induced production of prostaglandin (PGE)2 by pulmonary macrophages. PGE2 has crucial down-regulatory effects on fibroblasts including decreased fibroblast proliferation and decreased matrix production by fibroblasts. Regulation of chemokine receptor expression is also crucial to appropriate pulmonary repair. Fluorescein-isothiocyanate (FITC)-induced pulmonary fibrosis is regulated by MCP‐1 and CCR2 expression 154. FITC challenge results in the generation of MCP‐1. MCP‐1 signalling via the CCR2 receptor results in the generation of pro-fibrotic signals. Mice deficient for CCR2 are protected from the fibrotic response. The protection from fibrosis was relatively specific to the deletion of the CCR2 gene as mice with the deletion in the CCR5 gene were not protected from FITC-induced pulmonary fibrosis. Thus, a crucial role for CCR2 and its ligand(s) exists during the generation of a fibrotic response to lung injury.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fibroproliferative changes in fibrotic lung diseases. Altered growth factor production during fibroproliferative responses leads to an altered fibroblast phenotype which favours matrix deposition. Epithelial cell growth factors are importantly involved in regulating epithelial cell repair. Growth factors play crucial roles in regulating rates of apoptosis. Chemokines play crucial roles in regulating neo-vascularization during the repair process. IL: interleukin; LPS: lipopolysaccharide; IFN: interferon; GM-CSF: granulocyte macrophage colony stimulating factor; Th: T‐helper cell; NK: natural killer; MIP: macrophage inflammatory protein; MCP: monocyte chemoattractant protein.
Chemokines and their receptors also play a central role in regulating angiogenesis, a central biological event in repair and remodelling. IL‐8 and ENA-78 are CXC chemokines that contain the sequence GLULEU-ARG (ELR motif). ELR motif-containing CXC chemokines are potent angiogenic factors. In contrast, platelet factor‐4 and IFN‐γ inducible protein-10 are CXC chemokines that lack the ELR motif. Non-ELR containing chemokines can inhibit angiogenic activity of both ELR-CXC chemokines and structurally unrelated macrophage-derived angiogenic factor-bFGF 155.
Attributes of the pulmonary-cytokine receptor interaction
The molecular dialogue between cytokines and their receptors has the attributes of language, namely abstraction, combinatorial signals, semantics, syntax and context 156. Cytokines serve as abstractions of another reality. Cytokines report the presence of microbes and injured epithelia. Combinatorial signals make it possible to use a limited number of elements to transmit a large number of signals. Cytokines can be combined in many different patterns to create numerous, very complex messages. Semantics is the study of relationships between a sign and its meaning. The meaning of the sign can be defined operationally by the response that the sign induces. Cytokines in this context clearly have meaning since they induce measurable effects. Syntax refers to the organized structure of a string of signals; the grammar of the message. Cytokine signals, accordingly, have a syntax; the order in which cytokine signals are delivered is clearly important to the message. Finally, cytokines create a context. The milieu that surrounds a signal can clearly determine the nature of the response that it elicits. The meaning of an antigen, the nature of the response it elicits, depends greatly on the cytokine context in which the antigen appears. A T1 mix of cytokines predicates a very different response to a given antigen than does a T2 mix of cytokines.
Cytokines and their receptors comprise a “language” that is crucial to cellular function. Cell-cell communications proceed as an ongoing chemical dialogue that functionally connects recognition with responses. Understanding this language should permit the use of its component parts as tools to manipulate cellular behaviour in various clinical settings.
- Received July 31, 2001.
- Accepted August 1, 2001.
- © ERS Journals Ltd