





Abstract
Rationale The SARS-CoV-2/COVID-19 pandemic has highlighted the serious unmet need for effective therapies that reduce ARDS mortality. We explored whether extracellular nicotinamide phosphoribosyltransferase (eNAMPT), a ligand for Toll-like receptor 4 and a master regulator of innate immunity and inflammation, is a potential ARDS therapeutic target.
Methods Wild type C57BL/6J or endothelial cell (EC)-cNAMPT−/− knockout mice (targeted EC NAMPT deletion) were exposed to either a lipopolysaccharide (LPS)-induced (“one-hit”) or a combined LPS/ventilator (“two-hit”)-induced acute inflammatory lung injury model. A NAMPT-specific mAb imaging probe (99mTc-ProNamptorTM) was used to detect NAMPT expression in lung tissues. Either an eNAMPT-neutralising goat polyclonal antibody (pAb) or a humanised monoclonal antibody (ALT-100 mAb) were utilised in vitro and in vivo.
Results Immunohistochemical, biochemical, and imaging studies validated time-dependent increases in NAMPT lung tissue expression in both preclinical ARDS models. Intravenous delivery of either eNAMPT-neutralising pAb/mAb significantly attenuated inflammatory lung injury (H & E staining, BAL protein, BAL PMNs, plasma IL-6) in both preclinical models. In vitro human lung EC studies demonstrated eNAMPT-neutralising antibodies (pAb, mAb) to strongly abrogate eNAMPT-induced TLR4 pathway activation and EC barrier disruption. In vivo studies in wild type and EC-cNAMPT−/− mice confirmed a highly significant contribution of EC-derived NAMPT to the severity of inflammatory lung injury in both preclinical ARDS models.
Conclusions These findings highlight both the role of EC-derived eNAMPT and the potential for biologic targeting of the eNAMPT/TLR4 inflammatory pathway. In combination with predictive eNAMPT biomarker and NAMPT genotyping assays, this offers the opportunity to identify high-risk ARDS subjects for delivery of personalised medicine.
Abstract
Underscoring the therapeutic potential for targeting the eNAMPT/TLR4 pathway in ARDS/VILI, a humanised eNAMPT-neutralising monoclonal antibody (mAb) was highly effective in reducing the severity of ARDS in these dual complementary preclinical ARDS models.
INTRODUCTION
The SARS-CoV-2/COVID-19 pandemic and the unprecedented numbers of deaths due to COVID-19-induced acute respiratory distress syndrome (ARDS) have dramatically highlighted multiple unmet needs for ARDS patients. These include the absence of validated ARDS biomarkers and the absence of effective FDA–approved ARDS pharmacotherapies to address the associated lethal multi-organ failure. Although insights into ARD/VILI pathobiology are limited, a key advance has been the appreciation of bacteria- and virus-induced activation of evolutionarily-conserved systemic inflammatory networks [1–3], releasing a “cytokine storm” that increases lung and systemic vascular permeability, organ edema, and multi-organ dysfunction [4–7] ultimately increasing COVID-19-ARDS and non-COVID-19 ARDS mortality [8–10]. The life-threatening organ dysfunction is caused by dysregulated host responses to infection [6] mediated via interactions of pathogen-associated molecular patterns (PAMPs) and pattern recognition receptors (PRRs) that can also be activated by host nuclear, mitochondrial, and cytosolic proteins known as damage-associated molecular patterns (DAMPs). DAMPs are also released in response to danger signals such as hypoxia, cancer, trauma, and inhalation injury, thus potentially perpetuating a non-infectious inflammatory response [11].
We previously utilised preclinical multi-specie ARDS models coupled with genomic–intensive approaches [12, 13] to identify novel ARDS biomarkers and targetable pathways [12–18] and identified nicotinamide phosphoribosyltransferase (NAMPT) as a novel DAMP [19] and attractive ARDS target. We showed NAMPT expression to be highly induced by multiple ARDS-related stimuli including bacterial infection, hypoxia, shock, trauma, and excessive mechanical stress produced by mechanical ventilation [20–23]. Reduced expression of the gene encoding NAMPT (NAMPT) via siRNAs, miRNAs, and utilisation of NAMPT+/− heterozygous mice, dramatically attenuated the severity of preclinical ARDS/VILI injury [17].
NAMPT is a cytozyme whose intracellular enzymatic activities (iNAMPT) regulate nicotinamide adenine dinucleotide (NAD) biosynthesis [24, 25], contributing to injury in a tissue- and cell-specific manner [26]. Our studies, however, much more strongly implicate secreted extracellular eNAMPT as the primary mechanism of NAMPT's contribution to inflammatory lung inury and the increased mortality of critically ill ARDS patients on mechanical ventilation [17, 19, 27]. eNAMPT is a master regulator of inflammatory networks via binding to the PRR, Toll-like receptor 4 (TLR4) [19], eliciting profound NFkB-mediated inflammatory cytokine release that increases vascular permeability and multi-organ dysfunction directly linked to ARDS mortality [14, 28–30]. Two critical observations link NAMPT expression and function to human ARDS pathobiology. First, eNAMPT plasma levels alone [14, 31–33], or as part of an ARDS plasma biomarker panel [14, 31–33], are associated with ARDS severity and mortality. Secondly, NAMPT genetic variants that alter promoter activity in response to excessive mechanical stress [14, 22, 29, 30] and hypoxia [23] also confer increased ARDS susceptibility and mortality [14, 22, 29, 30].
In the current study, we explored the validation of eNAMPT as a viable ARDS therapeutic target in “one-hit” (LPS) and “two-hit” (LPS/VILI) preclinical ARDS models. Intravenous administration of eNAMPT-neutralising antibodies, either a polyclonal (pAb) or a humanised monoclonal (mAb), significantly reduced the severity of murine acute inflammatory lung injury. Our study also confirms the critical role of endothelial cell (EC)-derived eNAMPT in ARDS pathobiology, extending previous reports of robust spatially-localized NAMPT expression in lung endothelium, epithelium, and resident and infiltrating leukocytes but without assessment of cell-specific NAMPT contributions to ARDS severity. Utilising EC-specific conditional NAMPT KO mice, we now show the significant and unequivocal involvement of EC-secreted eNAMPT in dual preclinical ARDS models of injury. These studies strongly validate the viability of eNAMPT as an ARDS therapeutic target and underscore the capacity of an eNAMPT-neutralising biologic therapy to directly address the unmet need for novel strategies that improve ARDS/VILI mortality.
MATERIALS AND METHODS
Reagents
Recombinant human eNAMPT was purchased from Peprotech (Cranbury, NJ). Antibodies that are immunoreactive against p-NFkB, pp-ERK, pp-p38, pp-JNK, IL-6, and IL-8 (KC) were purchased from Cell Signalling Technologies (Danvers, MA) and against β-actin from Invitrogen (Carlsbad, CA) (NFkB). Goat, rabbit, and mouse secondary antibodies were purchased from Life Technologies (Waltham, MA). IgG for use as controls was obtained from Jackson ImmuneResearch (West Grove, PA). Goat anti-human NAMPT pAb was custom-generated as previously described [17]. All other reagents were from Sigma-Aldrich (St. Louis, MO).
Generation of an eNAMPT-neutralising humanised mAb
Two eNAMPT-neutraliding humanised mAbs, ALT-100 and ALT-300 were provided by Aqualung Therapeutics Corporation (Tucson, AZ) following in vitro mAb screening utilising trans-EC electrical resistance assays [34, 35], NFkB activation biochemical assays [19] and in vivo screening utilising dual preclinical ARDS models. ALT-100 was selected as the lead in vivo therapeutic and ALT-300 chosen for incorporation into the tissue NAMPT-imaging probe, 99mTc-ProNamptorTM. Details of mAb generation and selection are supplied in Supplemental Materials and Methods.
99mTc-ProNamptorTM mAb imaging
Extremely high NAMPT protein sequence homology exists between mice, rats, non-human primates, and humans (95–99%), underscoring ALT-100 and ALT-300 mAb utility in the murine preclinical studies we conducted including the ALT-300 mAb-containing fluorescent and 99mTc-labeled probes used for tissue imaging of NAMPT expression with human IgG serving as control [36, 37] (details of ALT-300 Cy5.5 or 99mTc labelling are available in Supplemental Materials and Methods). A mouse model of skin inflammation [38, 39] induced by topical application of 12-O-tetradecanoylphorbol-13-acetate (TPA) was utilised to validate the ability of the Cy5.5-ALT-300 probe to detect NAMPT tissue expression in vivo. TPA was applied to the surface of the right ear, reapplied at 24 h, and the left ear treated with acetone as the negative control. In separate experiments (n=3 mice), either Cy5.5-ALT-300 or Cy5.5-IgG (15–20 µg) was injected intravenously 3 h after the second TPA/PBS application, followed by mouse imaging. To assess the capacity of the 99mTc-ALT-300 probe to detect in vivo NAMPT tissue expression, 99mTc-ProNamptorTM (1.0–1.5 mCi, >98% radiochemical purity) [40, 41], or IgG control Ab was intravenously injected at 3 h post LPS challenge in the “one-hit” model and imaged (Quantum Imaging Detector camera) [42–44]. Count activity-based measurements of 99mTc-ProNamptorTM biodistribution and ex vivo autoradiography were performed in harvested lungs (see Supplemental Materials and Methods for additional details).
Mouse strains
In vivo experiments utilised either wild type male C57BL/6J mice (8–12 weeks, Jackson Laboratories, Bar Harbor, ME), EC-specific conditional NAMPT knockout mice (EC-cNAMPT−/−) on a mixed 129/B6 background, or littermate NAMPTfl/fl controls. EC-cNAMPT−/− mice were generated by crossing floxed NAMPT mice (NAMPTfl/fl) with tamoxifen-inducible EC-specific Cre transgenic mice (Tek-Cre/ERT2-1Soff) [45] with backcrossing with floxed homozygous NAMPT mice. After the final dose of tamoxifen, a 2 weeks minimal wait period was implemented before the utilisation of EC-cNAMPT−/− mice for experimentation (see Supplemental Materials and Methods for additional details). EC-specific KO mice carrying the NAMPT flox transgene did not display discernible differences in phenotypic traits compared to their wild-type littermates. Growth rate, fecundity, and fertility also did not differ from wild-type mice. Similarly, NAMPT flox mice crossed with the TIE2/ERT2 Cre mice to generate the conditional NAMPT KO line did not exhibit any phenotypic differences from either littermates or parental strains, both before and after tamoxifen injections.
eNAMPT-neutralising strategies in “one-hit” and “two-hit” preclinical ARDS models
For the “one-hit” ARDS model, mice received an intratracheal LPS injection and were sacrificed at 18 h post LPS as previously described [46–48]. For the “two-hit” LPS/VILI ARDS model, similar LPS-exposed mice were reintubated at 18 h and placed on mechanical ventilation for 4 h (tidal volume 20 mL·kg−1, respiratory rate 90 breaths·min−1, positive-end expiratory pressure 0 cm H2O) as previously described [46–48]. In specific experiments, C57BL/6J mice received either the IV delivered eNAMPT-neutralising pAb or the ALT-100 mAb (4 mg·kg−1 and 0.4 mg·kg−1, respectively) or IgG control Ab (4 mg·kg−1) (see Supplemental Materials and Methods for additional details).
Bronchoalveolar lavage (BAL) analysis
BAL fluid retrieval, protein analysis, and cell count analysis including PMN determinations were performed as previously described [49] with additional details provided in the Supplemental Materials and Methods.
Evans Bue Dye extravasation assay
We evaluated lung vascular leakage by measuring extravascular Evans Blue Dye in the lung as we have rpeviousy described (58). Briefly, mice were injected Evans blue dye (0.05 mg, Sigma) i.v. 60 min before euthanasia. Lungs were then perfused to remove the intravascular dye, excised and homogenised in PBS. One volume of lung homogenate was incubated with 2 volumes of formamide and incubated at 60°C for 18 h before centrifugation. The optical density of the supernatant was measured at 620 nm and 740 nm using an Imark microplate reader. Concentrations of Evans blue were corrected for the presence of heme pigments using the following formula: A620 (corrected)=A620 (raw)—(1.1927×A720)+0.0071. The extravasated Evans blue dye concentrations were then calculated against a standard linear curve as a reflection of vascular protein leak into lung tissues.
Quantitative lung histology and immunohistochemistry (IHC) analyses
Hematoxylin and eosin staining: Lungs were fixed and sectioned for routine H & E staining and imaging (Olympus digital camera, 10x magnification) [50]. NAMPT staining: The avidin-biotin-peroxidase method was utilised with a rabbit anti-human NAMPT pAb (1:1000 dilution, Bethyl Laboratories, Montgomery, TX) for IHC visualisation of NAMPT expression in lung tissues as previously described [14, 17, 19]. NAMPT/β actin/CD-31 co-staining in EC-cNAMPT−/− mice. Lung tissue sections from EC-cNAMPT−/− mice were incubated overnight (4°C) with primary rabbit anti-human NAMPT pAb (Bethyl), β actin or rat anti-CD-31 mAb and stained with biotinylated secondary antibody (1 h, 25°C) and imaged (Zeiss Axiovert Photomicroscope, 10X objective, NA 0.4). Quantitative analyses: Histological and IHC images were randomly selected for H & E or NAMPT quantification using ImageJ software [51] with additional details provided in the Supplemental Materials and Methods.
Acute Lung Injury Severity Score (ALISS) quantification
The Acute Lung Injury Severity Score (ALISS) was utilised to integrate lung injury indices in the “one-hit” and “two-hit” preclinical ARDS models and to standardise the injury levels across the in vivo models. A ranking point system, incorporating published recommendations [59] objectively assigns a score to each study animal (1 to 4 points) for each of 4 severity of injury readouts (H & E histology quantification, BAL total protein concentration, BAL total PMN cell count and plasma levels of the pro-inflammatory cytokine, IL-6). The maximal score for each animal is 16 points/mouse. In general, an ALISS score of 1–4 reflects the complete absence of injury, scores of 5–8 points reflect mild injury, scores of 9–12 points reflect moderate injury, and scores >12 points reflect severe injury. Additional details are provided in the Supplemental Materials and Methods.
Trans-endothelial electrical resistance (TER) measurements
Human pulmonary artery EC (Lonza, Walkersville, MD) were cultured as previously described [52] and seeded on evaporated gold microelectrodes (37°C, 5% CO2) to measure TER using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described [53]. TER values from each microelectrode were pooled and plotted versus time (mean±sem).
Biochemical tissue and plasma levels of eNAMPT, NFkB, MAP kinases, IL-6, and IL-8
Western blotting of lung homogenates was performed according to standard protocols as previously reported [23, 49] with densitometric quantification of lung tissue expression of NAMPT, NFkB, MAP kinases, IL-6, and βν-actin (total protein control). eNAMPT plasma levels were measured by ELISA as previously reported [27, 31], and plasma levels of IL-6 and IL-8 (KC) were measured utilising a meso-scale ELISA platform (Meso Scale Diagnostics, Rockville, MD) (described in Supplemental Materials and Methods).
Statistical analysis
Continuous data were compared using nonparametric methods and categorical data by the chi-square test. Where applicable, standard one-way ANOVA was used, and groups were compared using the Newman-Keuls test. Two-way ANOVA was used to compare the means of data from two or more different experimental groups. If a significant difference was present by ANOVA (p<0.05), the least significant differences (LSD) test was performed post hoc. Statistical tests were performed using GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com. Statistical significance was considered at p<0.05.
RESULTS
Increased lung tissue NAMPT expression in preclinical murine ARDS models
Compared to control mice, IHC lung tissue staining from mice exposed to either the “one-hit” (LPS 18 h) ARDS model (fig. 1a) or the “two-hit” (LPS 18 h, ventilation 4 h) ARDS/VILI model (fig. 1c) demonstrated dramatic upregulation of NAMPT expression (alveolar epithelium, endothelium, macrophages, neutrophils) summarised in figures 1b and d (p<0.05) and confirmed in lung homogenates from LPS-challenged mice. NAMPT protein expression was significantly elevated beginning 2 h post LPS challenge, peaking at 4 h, and returning towards baseline by 18 h (fig. 1e).

Increased NAMPT expression in lung tissues from “one-hit” and “two-hit” preclinical ARDS murine models. IHC staining to visualise NAMPT expression in lung tissues utilised a rabbit anti-human mAb (Bethyl, Montgomery TX). Compared to control, unchallenged C57BL/6J mice, marked increases in NAMPT expression were observed in lung tissues from mice exposed to either a “one-hit” LPS ARDS model (intratracheal LPS, 18 h) (a) or a “two-hit” preclinical ARDS/VILI model (LPS 22 h, ventilator exposure for final 4 h, tidal volume 20 mL·kg−1) (c). NAMPT expression was most prominent in alveolar epithelium, endothelium, macrophages, and infiltrating neutrophils (insets). Changes in NAMPT IHC staining were quantitatively summarised (Panels b and d, Image J software) (n>5/group, *p<0.05 for LPS or LPS/VILI versus control). We also assessed NAMPT protein expression in lung homogenates (Western blotting) at various times post LPS challenge (e). Densitometric analysis (n=4 each time point) showed significant time-dependent increases in NAMPT immunoreactivity, peaking at 4 h post LPS (*p< 0.05).
In vivo detection of NAMPT lung tissue expression
Validation of the ProNamptorTM mAb probe for detection of tissue inflammation was observed with the TPA-induced ear inflammation model with a significantly higher accumulation of Cy5.5-ProNamptorTM observed relative to the control Cy5.5-IgG (in vivo fluorescence imaging post-injection) (fig. 2a). In both unchallenged control mice and LPS-challenged mice exposed to injected either the 9mTc-IgG control or 99mTc-ProNamptorTM, there was prominent accumulation of radioactivity in the liver and in the cardiac blood pool and, to much lesser extent, in the lung (fig. 2b). However, in vivo and ex vivo imaging demonstrated significantly increased radioactive accumulation of 99mTc-ProNamptorTM in lung tissues from mice exposed to the “one-hit” LPS ARDS model (imaged at 7 h, fig. 2b) confirmed by ex vivo autoradiograph images (fig. 2c). Quantification of 99mTc uptake in lung tissues at 7 h post LPS challenge showed significant increases in both the non-specific IgG as well as ProNamptorTM, however, the magnitude of 99mTc-ProNamptorTM radioactivity uptake was significantly increased compared to IgG control and unchallenged mice (figs. 2c and d). The marked increase in 99mTc-ProNamptorTM radiolabel uptake at 7 h post LPS waned substantially by 10 h post LPS challenge (figs. 2d and e), a finding that is consistent with levels of NAMPT protein expression in lung tissue homogenates depicted in figure 1e.

ProNamptorTM detects NAMPT expression in vivo in the “one-hit” LPS preclinical ARDS model. a) To validate the eNAMPT-specific mAb-containing ProNamptorTM probe's capacity to detect inflammatory injury, studies were conducted in the TPA-induced inflammatory ear model. Representative fluorescence images of Cy5.5-IgG (left) and the eNAMPT-specific imaging probe, Cy5.5-ProNamptorTM (right), acquired 12 h after probe injection in two TPA-injected mice with right ear edema. Significantly enhanced fluorescence accumulation was observed in the inflamed right ear of a mouse injected with Cy5.5-ProNamptorTM compared to the Cy5.5-IgG-imaged mouse (arrows). b) Representative whole-body iQID images in an unchallenged control mouse and in LPS-challenged C57BL/6J mice injected with either a non-specific 99mTc-IgG Ab probe or with 99mTc-ProNamptorTM. Images were collected 7 h post LPS instillation (3 h post 99mTc-ProNamptorTM injection). Magnified thoracic images highlight the detection of NAMPT expression in LPS-induced lung inflammation. Increased radioactivity is noted for both the non-specific 99mTc-IgG Ab and the 99mTc-ProNamptorTM probes in the liver and the cardiac blood pool (outlined in red) of LPS-challenged animals. However increased lung accumulation was observed with the 99mTc-ProNamptorTM (chest area marked by white dashes), (c) Significantly higher radioactive accumulation of 99mTc-ProNamptorTM in LPS-injured lungs (7 h, compared to IgG Ab-99mTc and to control lungs) was further confirmed by ex vivo lung autoradiograph imaging (representative image, n=4). d and e) In vivo quantitative image analysis and ex vivo biodistribution measurements (%ID/g) demonstrate significant increased 99mTc-ProNamptorTM pulmonary uptake in LPS-injured lungs at 7 h post LPS with waning of NAMPT uptake/expression by 10 h compared to 99mTc-IgG Ab and control lungs (*p<0.01 compared to controls).
eNAMPT-neutralising strategies attenuate “one-hit” preclinical ARDS injury
H&E lung tissue staining from the “one-hit” LPS-challenged ARDS model show alveolar inflammation with significant neutrophil infiltration and alveolar edema compared to control mice (inset) (fig. 3a). Intravenous administration of either the eNAMPT-neutralising pAb (4 mg·kg−1) or the ALT-100 mAb (0.4 mg·kg−1) significantly reduced histologic injury (fig. 3a), which was verified by the significant reductions in LPS-induced inflammatory indices i.e. BAL protein (fig. 3c) and PMN counts (fig. 3d). The lung protection afforded by ALT-100 mAb was significantly superior to the eNAMPT pAb as reflected either by H&E staining (fig. 3b) or as captured in the integrated acute lung injury severity score or ALISS (H&E staining, BAL protein, BAL PMN counts, plasma IL-6 concentration) (fig. 3e).

eNAMPT-neutralising strategies attenuate “one-hit” preclinical ARDS injury. a) H&E lung tissue staining from mice exposed to the “one-hit” LPS/ARDS injury model (18 h, left) show interstitial and alveolar inflammation with significant neutrophil infiltration and alveolar edema compared to control C57BL/6J mice (inset). The severity of LPS-induced histologic injury is significantly reduced in mice receiving either an IV-administered eNAMPT-neutralising pAb (4 mg·kg−1, at time 0 with LPS injection) (center) or the humanised ALT-100 mAb (right) (0.4 mg·kg−1, at time 0 with LPS injection) (n=>5/group, *p<0.05 for LPS-Ab versus LPS-PBS control, **p<0.05 for LPS-mAb versus LPS-pAb). b) The ALT-100 mAb was significantly more effective than the eNAMPT pAb in reducing LPS-induced histologic evidence of inflammation and injury (*p<0.05 versus LPS-PBS, **p<0.05 mAb versus pAb). c and d) IV administration of either eNAMPT-neutralising biologic intervention (pAb or mAb) significantly reduced LPS-induced increases in BAL protein (c) and BAL PMN counts (d) (*p<0.05 versus LPS-PBS, **p<0.05 mAb versus pAb). e) The superior efficacy of the humanised ALT-100 mAb compared to the eNAMPT pAb was again verified in an Acute Lung Injury Severity Score (ALISS) comprised of H & E staining, BAL protein, BAL PMN counts and IL-6 plasma levels (*p<0.01 versus LPS-PBS, **p<0.01 mAb versus pAb).
eNAMPT-neutralising strategies attenuate “two-hit” ARDS/VILI injury
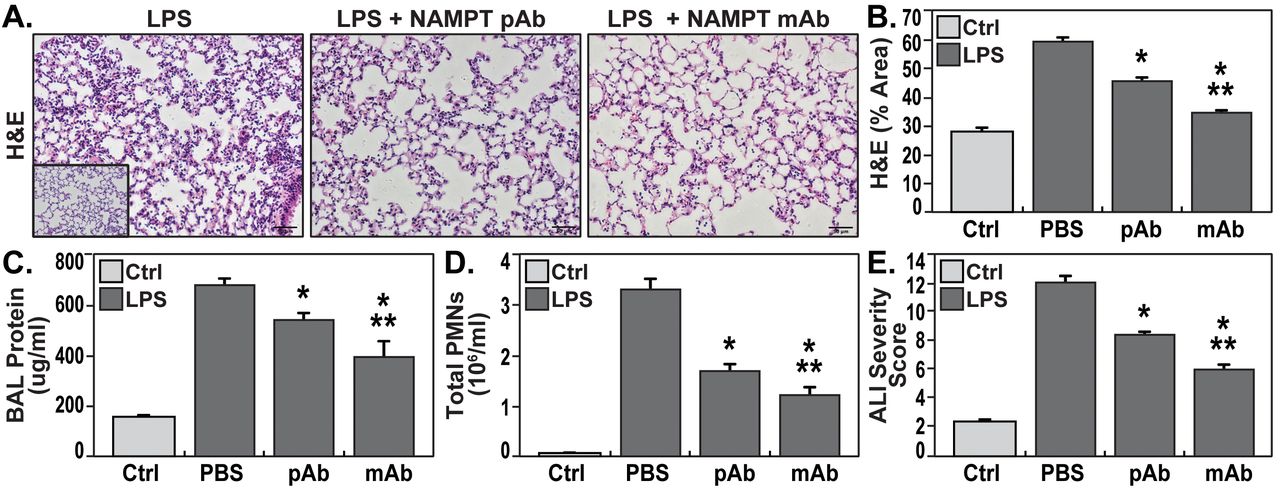
H&E staining in mice exposed to the “two-hit” ARDS/VILI model (LPS exposure 22 h, ventilator exposure for the final 4 h) revealed significant inflammatory cell infiltration into the lung parenchyma accompanied by both interstitial and alveolar edema when compared to control mice (inset) (fig. 4a). As with the “one-hit” LPS model, the severity of histologic injury in the “two-hit” LPS/VILI model was significantly reduced by i.v. delivery of either the eNAMPT-neutralising pAb (4 mg·kg−1) or the ALT-100 mAb (0.4 mg·kg−1) (figs. 4a and b). Both eNAMPT-neutralising biologic strategies also significantly reduced BAL protein (fig. 4c) and BAL PMN counts (fig. 4d) with greater ALT-100 mAb-mediated protection compared with the polyclonal Ab (fig. 4e).

eNAMPT-neutralising strategies attenuate “two-hit” preclinical ARDS/VILI injury. a) H&E lung tissue staining analysis in mice exposed to LPS (0.1 mg·kg−1,18 h) followed by mechanical ventilation (4 h, tidal volume 20 mL·kg−1) (left) were histologically compared to control C57BL/6J mice (inset) revealing significant parenchymal neutrophil infiltration and both interstitial and alveolar edema. The severity of LPS/VILI histologic injury was significantly reduced in mice receiving either the eNAMPT-neutralising polyclonal antibody (pAb, 4 mg·kg−1, at time 0 with LPS) (center) or the humanised ALT-100 mAb (0.4 mg·kg−1, at time 0 with LPS) (right). b) The ALT-100 mAb was significantly more effective than the pAb in reducing “two-hit” preclinical ARDS injury as captured by histologic quantification of injury (Image J software) (*p<0.05 versus LPS/VILI-PBS, **p<0.05 mAb versus pAb). c and d) Both eNAMPT-neutralising biologic interventions (pAb or mAb) also significantly reduced “two-hit” ARDS/VILI-induced increases in BAL protein content (c) and BAL PMN counts (d) after the “two-hit” LPS/VILI model (*p<0.05 versus LPS/VILI-PBS, **p<0.05 mAb versus pAb). e) Although both eNAMPT-neutralising biologic interventions (pAb, mAb) significant reduced LPS/VILI inflammatory injury, the ALT-100 mAb proved more effective than the eNAMPT pAb as verified in the ALISS (n=5 mice/group) (*p<0.05 versus LPS/VILI-PBS, **p<0.05 mAb versus pAb).
The eNAMPT neutralisation attenuates LPS- and LPS/VILI-challenged human lung EC signalling and barrier responses in vitro and in vivo
We previously demonstrated that NAMPT expression in a preclinical VILI model is primarily spatially-localized to lung EC, lung epithelium, and to resident and infiltrating leukocytes [13, 14]. We sought to specifically characterise lung endothelium as a target tissue for circulating eNAMPT and to assess the contribution of lung EC-derived NAMPT to the severity of acute lung injury in preclinical models of ARDS/VILI. Initial in vitro experiments utilising human lung EC confirmed that eNAMPT ligation of TLR4 induces robust NFkB phosphorylation [19] and MAP kinase activation (p38, JNK, p42/44 ERK) with these signalling responses nearly abolished by the eNAMPT neutralising pAb (figs. 5a and b) as were eNAMPT-induced declines in trans-human lung EC electrical resistance (TER), reflecting pAb- and mAb-mediated protection against eNAMPT-induced loss of EC barrier integrity (figs. 5c and d). The ALT-100 mAb, but not the eNAMPT-neutralising pAb, also produced significant reductions in LPS-induced barrier disruption (fig. 5d), results consistent with the contributory role of LPS-mediated eNAMPT secretion (maximal at 4 h, fig. 1e) in loss of EC barrier integrity and reductions in TER.

eNAMPT-neutralising strategies attenuate eNAMPT-induced human lung EC signalling and barrier responses. a and b) Human lung ECs were challenged with either human recombinant eNAMPT alone (1 ug·mL−1, 1 h) or a eNAMPT-pAb antibody mixture (eNAMPT 1 ug·mL−1+pAb 10 ug·mL−1, 1 h). Cells were next lysed and probed for phospho-proteins and total β-actin via Western Blot. eNAMPT-induced robust phosphorylation of NFKB, in addition to evidence of MAP kinase activation (pp-p38, pp-JNK, pp-42/44 ERK). Heat-denatured (100°C, 5 min) human recombinant eNAMPT (HD) (1 μg·mL−1, 1 h) served as a negative control confirming that eNAMPT effects do not reflect endotoxin contamination. The addition of the eNAMPT-neutralising pAb nearly totally abolished eNAMPT-induced NFkB phosphorylation and inhibited eNAMPT-induced MAP kinase activation detected by phosphorylation of ERK, p38, and JNK MAP kinases captured by densitometric measurements (n=3). c) In companion experiments, human lung ECs plated onto gold microelectrodes were challenged with either recombinant human eNAMPT alone (1 µg·mL−1), an eNAMPT-pAb mixture (eNAMPT 1 ug·mL−1 and pAb 10 ug·mL−1) or an eNAMPT-ALT-100 mAb mixture (eNAMPT 1 ug·mL−1 and ALT-100 10 ug·mL−1). Both eNAMPT-neutralising strategies, pAb, and mAb, attenuated eNAMPT-induced declines in EC barrier integrity compared to eNAMPT alone. Human lung ECs on gold microelectrodes were also challenged with LPS (1 µg·mL−1), with either PBS, eNAMPT pAb (10 µg·mL−1) or ALT-100 mAb (10 µg·mL−1) added immediately after LPS stimulation. The ALT-100 mAb, but eNAMPT pAb, also produced significant reductions in LPS-induced declines in EC barrier integrity compared to LPS alone. For TER studies, normalised resistance values >1 indicate lung EC barrier enhancement, normalised resistance values <1 indicate lung EC barrier disruption. d) Bar graph quantification of the TER declines and loss of barrier integrity, where data are expressed as change in TER compared to normalised unstimulated controls at 6 h (± sem, n=3 independent experiments per condition, *p<0.05 agonist alone versus agonist-pAb or mAb). e) Lung tissue homogenates from “one-hit” LPS-challenged (1 mg·kg−1, 18 h) with and without treatment with the eNAMPT-neutralising mAb (0.4 mg·kg−1 at time 0 h) were evaluated for Evans Blue Dye accumulation in lung tissues as a reflection of extravascular dye leakage and reported as the EBD concentration (μg/gram lung tissue) [58]. The bar graph demonstrates that the significant LPS-induced increases in EBD accumulation are abolished by prior addition of the ALT-10 mAb (*p<0.05 LPS versus LPS+mAb).
The contribution of circulating eNAMPT to the loss of lung vascular barrier integrity was next examined in vivo initially with the Evan's blue dye accumulation in lung tissues, an index of vascular permeability, which demonstrated dramatic ALT-100 mAb-mediated restoration of EC barrier integrity in the “one-hit” (LPS) preclinical ARDS model (fig. 5e). To validate our in vitro findings that specifically characterised increased MAP kinase signalling in eNAMPT-challenged lung endothelium, we examined levels of MAP kinase pathway signalling in lung tissues from mice exposed to both “one-hit” and “two-hit” preclinical ARDS models. Figure 6 demonstrates the activation of NFkB and MAP kinase family effectors (p38, JNK, p42/44 ERK), reflected by phosphorylation in lung tissue homogenates [19], in both preclinical ARDS models. Importantly, the ALT-100 mAb effectively suppressed the robust NFkB and MAP kinase family activation observed in the two ARDS models consistent with the contribution of circulating eNAMPT to the severity of lung injury in preclinical models of ARDS/VILI (fig. 6).
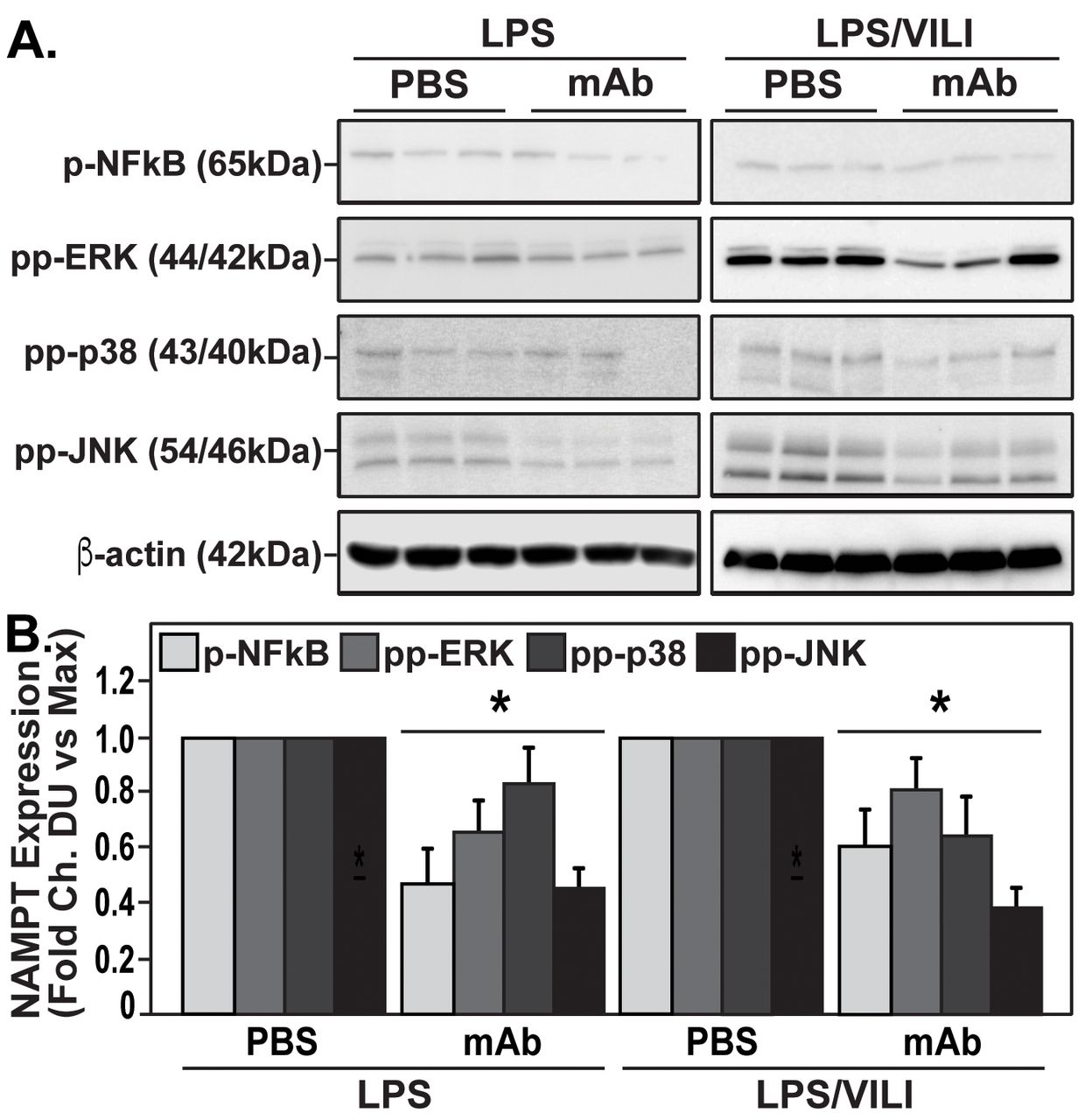
The eNAMPT-neutralising ALT-100 mAb attenuates MAP kinase signalling in “one-hit” and “two-hit” preclinical ARDS/VILI models. a) Lung tissue homogenates from “one-hit” LPS-exposed (1 mg·kg−1, 18 h) and “two-hit” LPS/VILI-exposed (LPS 22 h; mechanical ventilation 4 h, VT 20 mL·kg−1) C57B6 wild type mice were probed for phospho-proteins and total β-actin (Western Blot). LPS- and LPS/VILI-challenged mice displayed robust NFkB and MAP kinase phosphorylation (pp-p38, pp-JNK, pp-42/44 ERK) as evidence of pathway activation. Treatment with the eNAMPT-neutralisingALT-100 mAb (0.4 mg·kg−1, at time 0 h) resulted in signifiant reductions in both NFkB and MAP kinase pathway activation in the “one-hit”- and “two-hit” exposed mice captured by densitometric measurements (b) (n=3).
EC-specific NAMPT deletion in vivo reduces “one-hit” and “two-hit” lung injury
We utilised genetically-engineered conditional EC-cNAMPT−/− KO mice, with conditional NAMPT deletion restricted to EC, to further explore the contribution of lung EC-specific expression and secretion of NAMPT to the development of “one-hit” and “two-hit” lung injury. Lung tissue IHC studies demonstrated that tamoxifen-treated EC-cNAMPT−/− KO mice exhibit selective loss of NAMPT staining in lung endothelium whereas lung epithelial NAMPT expression was robust (fig. 7a), reflecting conditional deletion of NAMPT expression in lung EC (compared to wild type mice). Examination of H&E lung tissue staining in “one-hit” or “two-hit”-exposed EC-cNAMPT−/− KO mice showed significant reductions in inflammatory lung tissue injury (figs. 7b–d), accompanied by reduced BAL protein levels (fig. 7e) and BAL PMN counts (fig. 7f) compared to similarly exposed littermate control mice. The significant protection afforded by EC-specific NAMPT deletion in EC-cNAMPT−/− mice was captured in the integrated lung injury severity score (ALISS, fig. 7g).

EC-cNAMPT−/− mice are significantly protected from lung injury in “one-hit” and “two-hit” ARDS injury models. a) Lung tissue sections were obtained from EC-cNAMPT−/− mice sacrificed >2 weeks post-initiation of tamoxifen treatment (75 mg IP daily for 1 week) and tissue slides prepared for dual IHC staining with cell-specific double staining fluorescent-tagged NAMPT (green) and actin or CD-31 (red). In contrast to the dual staining in lung endothelium in littermate control sections (colocalized merge, left), tamoxifen-treated EC-cNAMPT−/− mice exhibit an absence of NAMPT staining in vascular cells while NAMPT staining in lung epithelium and leukocytes is prominent consistent with our original report [14]. These results reflect the targeted NAMPT deletion and absent expression in vascular endothelium. b–d) H&E staining of WT mice and EC-cNAMPT−/− mice exposed to the “one-hit” LPS ARDS model (b) or to the “two-hit” LPS/VILI model (c) demonstrates significantly reduced inflammatory lung injury in mice with conditional deletion of EC NAMPT, summarised by Image J quantification (d). e–g) “One-hit” LPS- and “two-hit” LPS/VILI-exposed EC-cNAMPT−/− mice also exhibit significant reductions in BAL protein levels (e) and BAL PMN counts (f) with this protection captured in the integrated ALISS score (g). *p<0.05 EC-cNAMPT−/− versus similarly-exposed littermates.
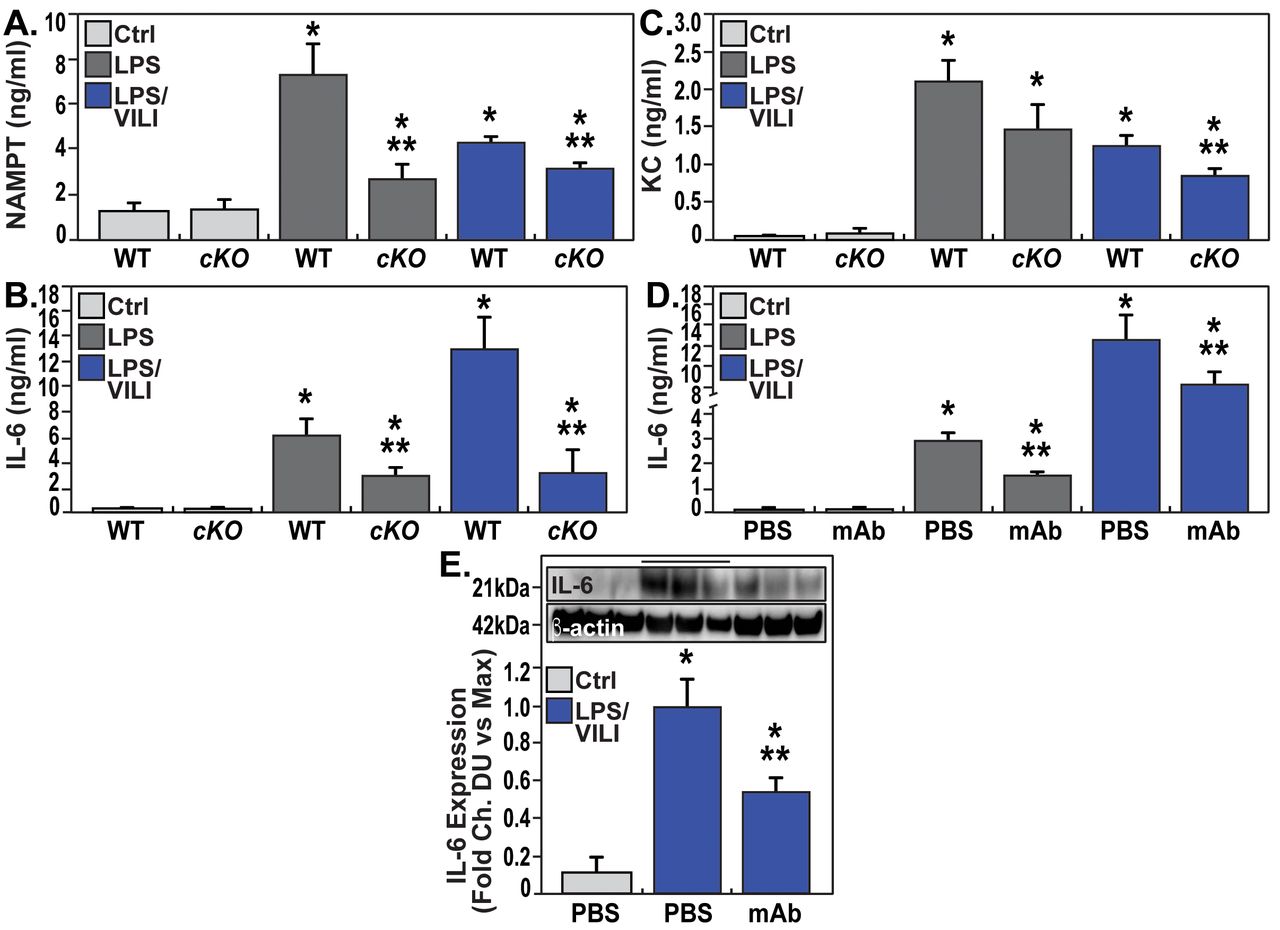
Lastly, we explored the involvement of EC-derived circulating eNAMPT to the severity of acute lung injury and measured eNAMPT plasma levels in “one-hit”- and “two-hit”-exposed EC-cNAMPT−/− and littermate control mice. Compared to littermate controls, both “one-hit”- and “two-hit”-exposed EC-cNAMPT−/− mice demonstrated reduced plasma eNAMPT levels (fig. 8a) as well as reduced plasma levels of IL-6 and IL-8 (KC in mice), two inflammatory cytokines often implicated as components in the ARDS “cytokine storm” (figs. 8b and c). Plasma levels of both IL-6 and IL-8 (KC) in “one-hit”- and “two-hit”-exposed WT C57Bl6 mice were also significantly reduced in ALT-100 mAb-treated mice compared to untreated mice (fig. 8d), validated by the decreased IL-6 protein expression in “two-hit” ARDS-exposed mice treated with the eNAMPT-neutralising ALT-100 mAb (fig. 8f).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EC NAMPT deletion and the ALT-100 mAb reduce plasma and tissue cytokine expression in “one-hit”- and “two-hit”-exposed ARDS mice. a) Plasma eNAMPT levels (ELISA) were significantly increased in “one-hit”- and “two-hit”-exposed littermate controls, with significantly reduced circulating eNAMPT levels in similarly exposed EC-cNAMPT−/− KO mice (*p<0.05 WT LPS or LPS-VILI versus WT Ctrl, **p<0.05 EC-cNAMPT−/− versus “one-hit”/“two-hit”-exposed littermates). b and c) Plasma levels of the murine inflammatory cytokines, IL-6 and KC (IL-8) in “one-hit”- and “two-hit”-exposed EC-cNAMPT−/− KO mice are reduced levels compared to similarly-exposed littermates. d) Treatment with the eNAMPT-neutralising humanised ALT-100 mAb reduces the increase in plasma levels of IL-6 in C57B6 wild type mice exposed to “one-hit” and “two-hit” preclinical ARDS injury models (*p<0.05 LPS or LPS/VILI alone versus Ctrl, **p<0.05 LPS or LPS/VILI mAb versus LPS or LPS/VILI alone). e) Western blot detection of IL-6 protein expression in lung tissue homogenates from “one-hit” LPS-exposed C57B6 wild type mice shows significant ALT-100 mAb-mediated reductions in IL-6 expression captured by densitometric measurements (n=4) (*p<0.05 LPS/VILI alone versus Ctrl, **p<0.05 LPS/VILI mAb versus LPS/VILI alone).
DISCUSSION
Currently, there are no FDA-approved ARDS therapies, a serious unmet need that's has been dramatically highlighted by the current COVID-19 pandemic. To potentially address the need for pharmacotherapies that reduce ARDS mortality, we previously identified eNAMPT, a novel DAMP [19], as a potentially attractive ARDS target whose expression and function tightly link to human ARDS. For example, NAMPT expression is highly induced by ARDS-relevant stimuli such as bacterial infection, hypoxia, shock, trauma and excessive mechanical stress produced by mechanical ventilation [20–23]. Further, plasma levels of eNAMPT are a biomarker for ARDS severity [14, 17, 22, 29–31, 33], and NAMPT genotypes with elevated minor allelic frequencies (i.e., common SNPs) confer a significant risk of increased ARDS severity and mortality in both Blacks and non-Hispanic whites [14, 17, 22, 29–31, 33]. Our earlier studies strongly implicated an essential contribution of eNAMPT to both VILI and ARDS pathobiology [14, 17, 22, 29–31, 33] via upstream activation of inflammatory cascades as a consequence of TLR4 ligation and NFkB transcriptional activities [19]. Thus, there is a substantial and compelling foundational basis for eNAMPT as a viable therapeutic target in ARDS/VILI.
Our current study underscores eNAMPT involvement in ARDS pathobiology in several important and novel ways. First, we utilised complementary approaches to validate increased NAMPT lung tissue expression in both “one-hit” (LPS) and “two-hit” (LPS/VILI) preclinical ARDS models. IHC and biochemical studies revealed markedly increased NAMPT lung tissue expression in both preclinical models. This was further verified utilising the radiolabeled probe, 99mTc-ProNamptorTM, with temporally increased LPS-induced NAMPT lung tissue expression that aligned with NAMPT expression in lung homogenates. As 99mTc-ProNamptorTM contains the radiolabeled eNAMPT-neutralising mAb (ALT-300), the potential exists for 99mTc-ProNamptorTM to serve as a novel molecular imaging theranostic that may therapeutically reduce inflammatory injury, a hypothesis to be addressed in future studies.
A second significant implication of this work is to extend our earlier studies in VILI murine models and directly explore eNAMPT-neutralisation as an effective ARDS therapy. Previous intratracheal instillation of an eNAMPT-neutralising polyclonal pAb supported eNAMPT as a relevant VILI therapeutic target [17, 19]. We now extend this work to demonstrate intravenous administration of ALT-100, the eNAMPT-neutralising humanised mAb, significantly reduced the severity of inflammatory lung injury in both preclinical ARDS models and was significantly more protective than the eNAMPT pAb. Our use of dual preclinical ARDS models adds significant rigor to our work and directly addresses suggestions that a major failure of ARDS pharmacologic interventions is due to use of a single preclinical ARDS murine model [54, 55].
Our preclinical results support our nascent ARDS clinical trial strategy which is designed to deliver the ALT-100 mAb to ARDS subjects with respiratory failure at the time of intubation i.e. prior to initiation of mechanical ventilation. This trial design specifically targets VILI prevention and the dampening of ventilator-induced amplification of inflammatory pathways to reduce multi-organ injury, thereby minimising the duration of mechanical ventilation, improving ICU survival, and reducing healthcare costs. Prior unsuccessful ARDS therapeutic clinical trials, primarily targeting single cytokines or bacterial products, were likely hampered by delays in targeted therapy delivery [56, 57], with initiation of therapy well after the onset of amplified innate immunity-driven lung and systemic inflammatory pathways. The feasibility of administering ALT-100 to ARDS subjects with impending respiratory failure in the ER or upon entry to the ICU, prior to intubation and exposure to mechanical ventilation, was previously a daunting challenge from a clinical trial perspective, but is supported by recent ER-ICU clinical trials.
Another valuable insight provided by our study is to underscore the critical importance of EC-derived circulating eNAMPT to ARDS pathobiology [14, 35]. While NAMPT expression, in addition to lung ECs, is robust in lung alveolar epithelium and in resident and infiltrating leukocytes in VILI-challenged canine and murine models [13, 14], the critical contribution of each cellular component to ARDS severity was previously unknown. Our current in vitro and in vivo studies confirm earlier work that highlighted lung EC as a key eNAMPT cellular target with dysregulation of lung EC permeability responses [35]. We now show potent eNAMPT-mediated activation of human lung EC TLR4 and MAP kinase pathways and loss of EC barrier integrity, responses strongly abrogated by both eNAMPT-neutralising modalities (pAb, ALT-100 mAb). The modest but significant inhibition of LPS-induced declines in trans EC electrical resistance barrier dysfunction by ALT-100 mAb is again consistent with the contribution of eNAMPT, secreted by EC in response to LPS, to increased vascular permeability, supported by the profound reductions in Evans blue dye acculumulation (fig. 5e) and dramatic reductions in MAP kinase signalling in vivo (fig. 6). To directly interrogate the role of EC-derived eNAMPT in vivo, we utilised an EC-cNAMPT−/− mouse line with targeted KO of eNAMPT that is conditionally restricted to the endothelium. EC-cNAMPT−/− mice were significantly protected in both “one-hit” and “two-hit” preclinical ARDS injury models and exhibited significantly reduced plasma levels of eNAMPT, IL-6, and IL-8 (KC) when compared to WT littermates. These results mirror the reductions in plasma and tissue levels of IL-6 in ALT-100-treated mice exposed to “one-hit” and “two-hit” ARDS models indicating that EC-derived circulating eNAMPT is likely a significant contributor to ARDS pathobiology.
In summary, our studies strongly validate targeting of the eNAMPT/TLR4 inflammatory pathway with a humanised eNAMPT-neutralising mAb as a viable therapeutic strategy to address the serious unmet need for effective and specific pharmacologic therapies to improve ARDS/VILI mortality. We have also clarified the essential and contributory role of EC-derived eNAMPT to inflammatory lung injury elicited by bacterial infection and by ventilator-induced mechanical stress. Finally, it is well known that the vast heterogeneity of ARDS has been a critical challenge to the conduct of successful therapeutic clinical trials in the U.S. and by ARDS clinical trial networks world-wide. Our study demonstrates that with the combined availability of i) eNAMPT as an ARDS predictive plasma biomarker, either alone [14, 32, 33], or as part of an ARDS biomarker panel [31]; ii) identification of high-risk NAMPT genotypes [14, 29, 30]; and iii) a highly efficacious eNAMPT-neutralising humanised mAb as targeted biologic therapy, the opportunity exists for novel ARDS clinical trial designs for that stratify patient enrollment for testing a biologic therapy that targets the eNAMPT/TLR4 inflammatory pathway, an attractive mechanism to deliver personalised ICU medicine in the current COVID-19 pandemic landscape.
Footnotes
Author Contributions: JGNG, SS – conception and design of the work, the analysis and interpretation of data for the work, the drafting and revision of the manuscript, approval of final version to be published; CB, AEC, ZL, DM – conception and design of the work, the analysis and interpretation of data for the work, critical revision of key intellectual content and approval of final version to be published. TB, SMC, ANG, CLK, HQ, DGV, JS – collection and analysis of data, revision of the manuscript, and approval of the final version to be published; CB, KB, JKB, AAD, SMD, EF, AG, JRJ, JM, LMV, VN, RCO, VRH, BS, XS – collected data and assisted with processing and manuscript revision
Support statement: This work was supported by the NIH/NHLBI grants P01HL126609, R01HL094394, and P01HL134610. National Heart, Lung, and Blood Institute; DOI: http://dx.doi.org/10.13039/100000050; Grant: P01HL126609, P01HL134610, R01HL094394.
Descriptor Number: 4.1 ALI/ARDS: Biological Mechanisms
Conflict of interest: Dr. Quijada has nothing to disclose.
Conflict of interest: Dr. Bermudez has nothing to disclose.
Conflict of interest: Dr. Kempf has nothing to disclose.
Conflict of interest: Mr. Valera has nothing to disclose.
Conflict of interest: Dr. Garcia has nothing to disclose.
Conflict of interest: Dr. Camp has nothing to disclose.
Conflict of interest: Dr. Song has nothing to disclose.
Conflict of interest: Dr. Franco has nothing to disclose.
Conflict of interest: Dr. Burt has nothing to disclose.
Conflict of interest: Dr. Sun has nothing to disclose.
Conflict of interest: Dr. Mascarenhas has nothing to disclose.
Conflict of interest: Dr. Burns has nothing to disclose.
Conflict of interest: Mr. Gaber has nothing to disclose.
Conflict of interest: Dr. OITA has nothing to disclose.
Conflict of interest: Reyes Hernon has nothing to disclose.
Conflict of interest: Dr. Barber has nothing to disclose.
Conflict of interest: Dr. Moreno-Vinasco has nothing to disclose.
Conflict of interest: Dr. SUN has nothing to disclose.
Conflict of interest: Dr. Cress has nothing to disclose.
Conflict of interest: Dr. Martin reports other from Aqualung, outside the submitted work;.
Conflict of interest: Dr. Liu has nothing to disclose.
Conflict of interest: NIH R01 (HL136603)- Ankit Desai
Conflict of interest: Dr. Natarajan has nothing to disclose.
Conflict of interest: Dr. Jacobson has nothing to disclose.
Conflict of interest: Dr. Dudek has nothing to disclose.
Conflict of interest: Dr. Bime has nothing to disclose.
Conflict of interest: Dr. Sammani has nothing to disclose.
Conflict of interest: Dr. Garcia reports grants and non-financial support from Aqualung Therapeutics, Corp, during the conduct of the study; grants and personal fees from Aqualung Therapeutics, Corp, outside the submitted work; In addition, Dr. Garcia has a patent US Patent No. 9,409,983 issued.
- Received June 27, 2020.
- Accepted November 5, 2020.
- Copyright ©ERS 2020
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










