





Coronavirus Disease 19 (COVID-19) is a novel emerging respiratory disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and is causing a vast pandemic with huge medical, social, personal and financial impact. Angiotensin-converting enzyme 2 (ACE2) was identified as the cell entry receptor used by SARS-CoV-2 [1, 2]. Importantly, smokers and patients with COPD are at increased risk for severe complications and a higher mortality upon SARS-CoV-2 infection [3]. We hypothesised that ACE2 expression is increased in lungs of smokers and patients with COPD, which may at least partially explain their higher risk for a more severe course of COVID-19. Therefore, we aimed to investigate the expression of ACE2 on both mRNA and protein level in a large number of lung tissue specimens of well phenotyped subjects, including never smokers, current smokers without airflow limitation, and patients with COPD.
In this cross-sectional observational study, we analysed lung tissue specimens from 134 subjects from our large lung tissue biobank at Ghent University Hospital and from explant lungs from end-stage COPD patients collected at UZ Gasthuisberg Leuven, Belgium. Ex-smoking was defined as smoking cessation for at least 1 year. COPD severity was defined according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification. Written informed consent was obtained from all subjects. This study was approved by the medical ethical committees of the Ghent University Hospital (2016/0132; 2019/0537) and the University Hospital Gasthuisberg Leuven (S51577).
RNA extraction from lung tissue blocks of 120 subjects was performed with the miRNeasy Mini kit (Qiagen, Hilden, Germany). Next, cDNA was prepared with the EvoScript Universal cDNA Master Kit (Roche), followed by RT-PCR analysis for ACE2 and 3 reference genes as described previously [4, 5].
Sections from formalin fixed paraffin embedded lung tissue blocks of 87 subjects were stained for ACE2. After antigen retrieval with citrate buffer (Scytek), the slides were incubated with anti-ACE2 antibody (polyclonal rabbit-anti-human, Abcam ab15248). Next, slides were colored with diaminobenzidine (Dako, Carpinteria, CA, USA) and counterstained with Mayer's hematoxylin (Sigma-Aldrich, St-Louis, MO, USA). Quantitative measurements of the ACE2-positive signal in alveolar tissue and bronchial epithelium were performed on images of stained paraffin sections as described previously [6].
Statistical analysis was performed with Sigma Stat software (SPSS 26.0, Chicago, IL, USA) and R3.5.1, using Kruskal-Wallis tests (on all 6 groups) followed by Mann-Whitney U tests (for the comparison between 2 groups), and multivariable linear regression analyses.
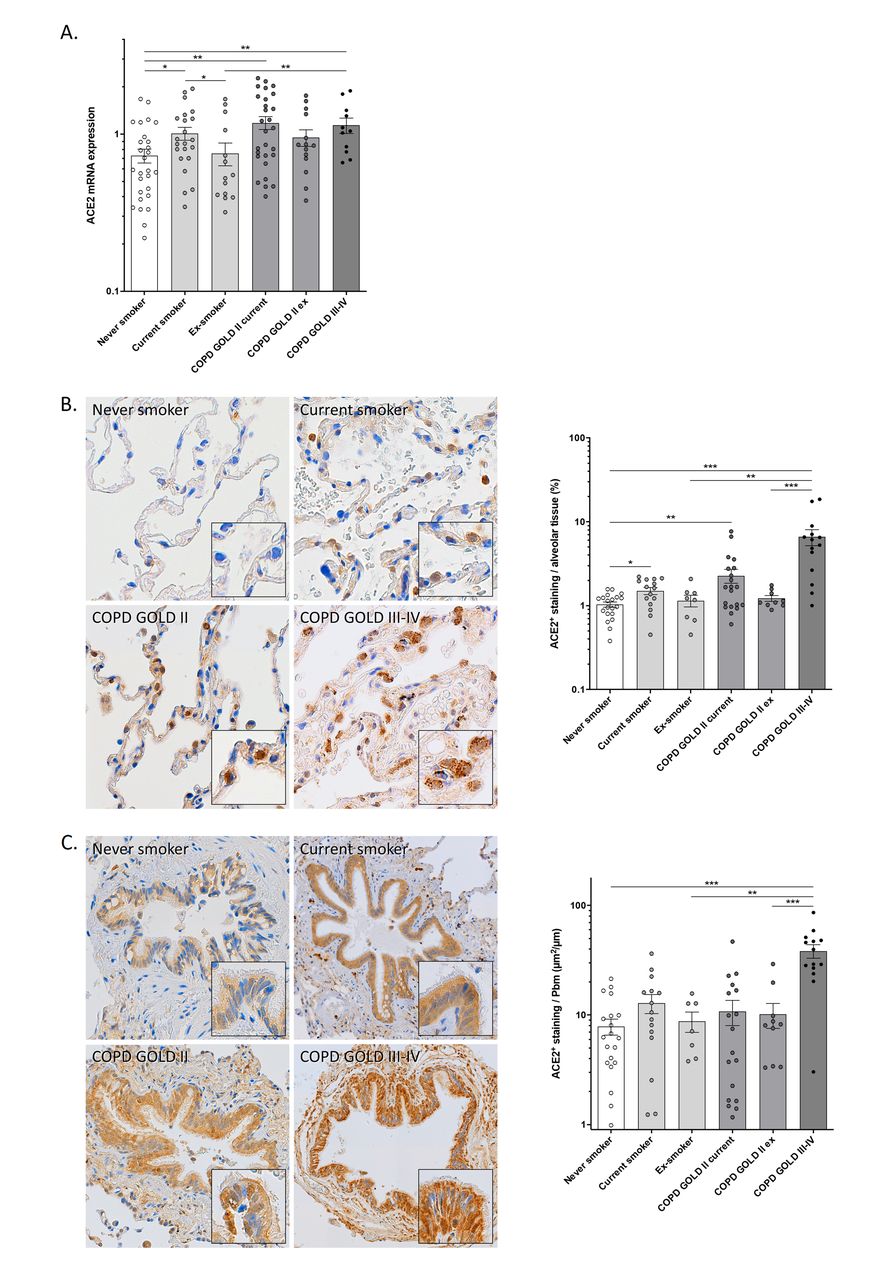
Using RT-PCR, ACE2 mRNA levels were determined in lung tissue from 120 subjects. ACE2 mRNA expression was significantly higher in lung tissue of current smokers without airflow limitation and current smokers with COPD (GOLD stages II and III-IV) compared to never smokers (fig. 1a). In addition, ex-smokers without airflow limitation showed significantly lower ACE2 mRNA levels, compared to current smokers. Multivariable linear regression analysis demonstrated that current smoking and COPD are both independently associated with increased ACE2 mRNA expression in lung tissue, even after adjustment for covariates including age, sex, BMI, and arterial hypertension (data not shown).

{kind=link}
Gene and protein expression of ACE2 in airways and lungs. a) ACE2 mRNA expression is increased in lung tissue of smokers and COPD subjects. Angiotensin-converting Enzyme-2 (ACE2) mRNA expression in lung tissue of never smokers, current and ex-smokers without airflow limitation and current and ex-smokers with moderate (GOLD II) or severe-to-very severe (GOLD III-IV) COPD, normalized to the expression of the housekeeping controls glyceraldehyde-3-phosphate dehydrogenase (GAPDH), peptidylprolyl isomerase A (PPIA) and succinate dehydrogenase complex flavoprotein subunit A (SDHA). Data are presented as means±sem. *p<0.05; **p<0.01. b) ACE2 protein levels are increased in alveolar tissue of smokers and COPD subjects. Representative images and quantification of ACE2 immunohistochemical staining in alveolar tissue of never smokers, smokers without airflow limitation, smokers with COPD GOLD stage II and smokers with COPD GOLD stage III-IV. The area of ACE2-positive signal was normalized to the total area of alveolar tissue present in each analyzed image. Data are presented as means±sem. *p<0.05; **p<0.01; ***p<0.001. c) ACE2 protein levels are increased in bronchial epithelium of smokers and COPD subjects. Representative images and quantification of ACE2 immunohistochemical staining in bronchial epithelium of never smokers, smokers without airflow limitation and smokers with COPD (GOLD stage II and III-IV). The area of ACE2-positive signal in each airway was normalized to the length of the basement membrane. Data are presented as means±sem. **p<0.01; ***p<0.001.
By immunohistochemical (IHC) staining, ACE2 protein levels were assessed in lung tissue from 87 subjects. ACE2 IHC revealed positive staining in both bronchial and alveolar epithelial cells, with the latter predominantly in alveolar type II cells (fig. 1b and c). Quantification of ACE2 protein levels in alveolar tissue revealed a significantly higher percentage of ACE2-positive alveolar tissue in current smokers without airflow limitation and current smokers with COPD (GOLD II and III-IV) compared to never smokers (fig. 1b). Moreover, the percentage of ACE2-positive alveolar tissue was significantly higher in patients with COPD GOLD III-IV, compared to ex-smokers without airflow limitation and ex-smokers with COPD GOLD II (fig. 1b). Quantification of ACE2 staining in bronchial epithelium revealed numerically higher levels in current smokers without airflow limitation and current smokers with COPD GOLD II, and significantly higher levels in patients with COPD GOLD III-IV, compared to never smokers (fig. 1c). Moreover, ACE2 protein levels in bronchial epithelium were significantly higher in patients with COPD GOLD III-IV, compared to ex-smokers without airflow limitation and ex-smokers with COPD GOLD II (fig. 1c). The observed association between COPD and ACE2 protein expression in alveolar tissue or bronchial epithelium remained significant after adjustment for possible confounders (data not shown).
As healthcare systems around the world are currently under great pressure due to the COVID-19 outbreak, identification of those at high risk is crucial. There is compelling evidence of a more severe course of COVID-19 in smokers and patients with comorbidities such as COPD. We clearly demonstrate an increased pulmonary expression of the SARS-CoV-2 entry receptor ACE2 in smokers and COPD subjects at both mRNA and protein level. While our observations complement previous reports on increased ACE2 mRNA and protein levels in whole lung tissue and bronchial epithelium [7–10], this is the first study demonstrating increased ACE2 protein in alveolar epithelium of smokers and patients with COPD, which can be directly linked to the site of injury when patients with severe COVID-19 develop dyspnea, hypoxia and pneumonia.
Currently, published data on COVID-19 in patients with COPD is fairly limited [11]. Nevertheless, an increased risk of developing severe COVID-19 as well as a higher mortality, has been reported in patients with COPD and in current smokers [3, 12]. Although there are several possible explanations for the increased susceptibility for severe COVID-19 in patients with COPD, including older age, comorbidities, dysregulated immune defenses and impaired mucociliary clearance, increased pulmonary expression of the SARS-CoV-2 entry receptor ACE2 is most likely another contributor [13]. Importantly, it has been demonstrated in mouse models that transgenic (over)expression of human ACE2 enhances the pathogenicity of SARS-CoV-1 and SARS-CoV-2 [14]. Moreover, human ACE2 was essential for viral replication in the lung.
The main strength of this study is the large number of lung tissue samples from well-phenotyped subjects that are included in the RT-PCR and IHC analyses. Moreover, IHC allowed us to quantify ACE2 protein levels in both bronchial and alveolar epithelium. However, certain limitations should be kept in mind. First, this study consists mainly of samples from lung resections for pulmonary tumors. This may introduce selection bias since altered expression of ACE2 in lung cancer has been suggested [15]. Second, samples of (very) severe COPD originate from a different patient population (lung transplantation), for which smoking cessation was an inclusion criterium. Finally, since we did not study lung tissue samples from COVID-19 positive subjects, we can only speculate on the importance of increased ACE2 in the pathogenicity of SARS-CoV-2.
In conclusion, we report higher ACE2 mRNA and protein levels in lung tissue of smokers and subjects with moderate to (very) severe COPD. Importantly, ACE2 protein levels are not only increased in bronchial but also in alveolar epithelium. Further research is needed to elucidate whether up-regulation of ACE2 expression in airways and lungs has consequences on infectivity and clinical outcomes of COVID-19.
Footnotes
Support statement: The research described in this article was supported by the Concerted Research Action of the Ghent University (BOF/GOA 01G00819) and by the Fund for Scientific Research in Flanders (FWO Vlaanderen, G052518N and EOS-contract G0G2318N). Fonds Wetenschappelijk Onderzoek; DOI: http://dx.doi.org/10.13039/501100003130; Grant: G052518N and EOS-contract G0G2318N; Universiteit Gent; DOI: http://dx.doi.org/10.13039/501100004385; Grant: BOF/GOA 01G00819.
Conflict of interest: Dr. Jacobs has nothing to disclose.
Conflict of interest: Dr. Van Eeckhoutte has nothing to disclose.
Conflict of interest: Dr. Wijnant has nothing to disclose.
Conflict of interest: Dr. JOOS reports grants from AstraZeneca, personal fees from Bayer, grants from Chiesi, personal fees from Eureca vzw, grants and personal fees from GlaxoSmithKline, personal fees from Teva, outside the submitted work; all fees were payed to the department.
Conflict of interest: Dr. Janssens reports grants from AstraZeneca and Chiesi.
Conflict of interest: Dr. Brusselle reports personal fees from Astra Zeneca, personal fees from Boehringer-Ingelheim, personal fees from Chiesi, personal fees from GlaxoSmithKline, personal fees from Novartis, personal fees from Sanofi, personal fees from Teva, outside the submitted work.
Conflict of interest: Dr. Bracke has nothing to disclose.
- Received May 26, 2020.
- Accepted June 19, 2020.
- Copyright ©ERS 2020
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.










