





Coronavirus disease 2019 (COVID-19) represents a public health crisis of pandemic proportions. Caused by SARS-CoV-2, which stands for severe acute respiratory syndrome (SARS) coronavirus 2, the symptoms most commonly reported include cough, fever, and shortness of breath, but also extra-pulmonary symptoms, such as neurological and gastroenterological manifestations.
The pathogenesis of the disease depends mainly on the mechanisms of entry and action of the coronaviruses. Until 2003, two types of coronavirus surface receptor were known. A number of group I coronaviruses, for example human coronavirus 229E and viruses causing transmissible gastroenteritis and feline infectious peritonitis, require the zinc metallo-protease aminopeptidase N (APN, CD13) for entry into their target cells [1, 2]. The group II coronavirus mouse hepatitis virus (MHV) uses members of the immunoglobulin superfamily of receptors such as the murine carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) [3]. Li and colleagues identified a distinct coronavirus as the etiological agent of the 2003 SARS [4], the SARS-CoV-1, which uses a surface glycoprotein called spike (S) to access host cells. Interestingly, SARS-CoV-2 is closely related to SARS-CoV-1, and it was demonstrated that S proteins of coronaviruses need to bind with cellular receptors to mediate infection of their target cells [5]. Li and coworkers were able to show that a metallopeptidase (angiotensin-converting enzyme 2, ACE2) [6], isolated from SARS-CoV-permissive Vero E6 cells (African green monkey kidney cell line), could efficiently bind the S1 domain of the SARS-CoV S protein and acted as a functional co-receptor for coronavirus entry. Most recently, ACE2 has been demonstrated to be a human interferon-stimulated gene suggesting that SARS-CoV-2 could exploit species-specific interferon-driven upregulation of ACE2, a tissue-protective mediator during lung injury, to enhance infection [7].
The disease pathogenesis also depends on the localisation of the coronavirus co-receptors. As shown by Hamming and colleagues, ACE2 is abundantly present in humans in the epithelia of lung and small intestine, cells in contact with the external environment, which might provide possible routes of entry for the SARS-CoV-2 [8]. This epithelial expression provides a first step in understanding the pathogenesis of the main SARS disease manifestations, in particular in the lung (cough, pneumonia and severe acute respiratory syndrome). Type I and type II pneumocytes are markedly positive for ACE2 indicating that alveolar pneumocytes are a possible site of entrance for SARS-CoV. Virus entry may cause cytopathological changes at the epithelial alveolo-capillary interface, initially resulting in induction of type II alveolar cells as a first attempt to repair. In SARS, the abundant expression of ACE2 in type II alveolar cells may cause rapid viral expansion and local alveolar wall destruction, resulting in rapidly progressive severe diffuse alveolar damage and hyperinflammation known as cytokine storm syndrome [9]. Moreover, it has been demonstrated that oxidative stress induced by SARS-CoV-2 can exacerbate DNA methylation defect, possibly resulting in further ACE2 demethylation and enhanced viremia [10]. Oxidative stress in the lung occurs when the antioxidant capacity is overwhelmed or depleted through external exposures, such as altered oxygen tension or air pollution, or internally by activation of resident cells or inflammatory cells recruited in response to an exposure, injury or infection [11, 12]. Interestingly, Abouhashem and coauthors have recently shown, through single cell RNA sequencing data of the human lungs, that specific components of the antioxidant defense system of the alveolar type II cells, such as superoxide dismutase (SOD)3 and activating transcription factor (ATF)4, an endoplasmic reticulum stress sensor, weaken in response to aging in the elderly donors. These results could contribute in part to the observed severity of COVID-19 in the elderly [13].
The other targets of SARS-CoV are immune organs and systemic small vessels, resulting in systemic vasculitis and decreased immune function. Other receptors/facilitators on the surface of human cells have been suggested to mediate the entry of SARS-CoV-2, including transmembrane serine protease 2 (TMPRSS2) [14], sialic acid [15] and extracellular matrix metalloproteinase inducer (CD147, also known as basigin) [16].
Interestingly, ACE2 as well as the other three facilitators are present in arterial and venous endothelial cells and arterial smooth muscle cells [8]. ACE2 is the most studied of these receptors/facilitators and questions have been recently raised about using renin-angiotensin-aldosterone system (RAAS) inhibitors, such as angiotensin II receptor blockers (ARB) and ACE inhibitors, for COVID-19 patients with hypertension [17–24]. In particular, data on a potential association between the use of ACE inhibitors or ARBs and the risk of developing SARS-CoV-2 were conflicting. Lately, two distinct studies have demonstrated, in large cohorts of patients, that there was no evidence of an increased risk of COVID-19 due to ACE inhibitor or ARB treatments [23, 25]. Of interest, it has also been shown that interleukin-6 produced during the cytokine storm syndrome can also be induced by angiotensin II through a mineralocorticoid receptor–dependent mechanism [26, 27].
The expression of ACE2 on endothelial, smooth muscle cells and perivascular pericytes in virtually all organs suggests that the SARS-CoV-2, once present in the circulation, can spread easily through the body [8]. Interestingly, in a recent work comparing post-mortem lung tissues from patients who died from COVID-19, acute respiratory distress syndrome (ARDS) due to influenza A(H1N1) infection and age-matched, uninfected control lungs, the authors found greater numbers of ACE2-positive endothelial cells and significant changes in endothelial morphology with disruption of intercellular junctions, cell swelling, and a loss of contact with the basal membrane [28].
Increasing clinical evidence shows that the most common co-morbidities observed in COVID-19 patients, that are associated with worse prognosis and higher rate of death, are systemic hypertension, diabetes and obesity, in which endothelial dysfunction [29] is known to be a key determinant. In this issue of the European Respiratory Journal, two research letters underscore the risk of deep vein thrombosis and acute pulmonary embolism in COVID-19 [30, 31]. These data confirm and extend previous observations supporting an underlying SARS-CoV-2-related endothelial dysfunction with an increased risk of venous thromboembolic disease, systemic vasculitis, endothelial cell apoptosis and inflammation in various organs [32–35]. It is also known that, as other infectious microorganisms, SARS-CoV-2 can activate coagulopathy through inflammatory responses. The discovery that platelet dense granules contain polyphosphates and secrete it upon activation led to the identification of extensive connections between the immune system and the coagulation cascade [36]. Polyphosphates released from activated platelets accelerate factor V activation, inhibit the anticoagulant activity of tissue factor pathway inhibitor, promote factor XI activation by thrombin, and contribute to the synthesis of thicker fibrin strands that are resistant in fibrinolysis [37]. The inflammatory effects of cytokines also result in activated vascular endothelial cells and endothelial injury with resultant prothrombotic properties. Vascular endothelial injury causes further thrombocytopenia, reduction of natural anticoagulants, but also hemostatic activation as the phenotypic expression of thrombotic diffuse intravascular coagulation. In the COVID-19 Associated Coagulopathy (CAC) [38], the mechanisms that activate coagulation in SARS-CoV-2 infection are still unknown but appear to be linked to inflammatory responses rather than specific properties of the virus [39].
Klok and coauthors have recently reported a 31% incidence of thrombotic complications in COVID-19 patients in intensive care unit [40]. Grillet et al. found a 23% of acute pulmonary embolism associated with COVID-19 pneumonia detected by pulmonary CT pulmonary angiography [41]. In addition, acute pulmonary edema can be observed in critical COVID-19 patients with occlusion and micro-thrombosis of small pulmonary vessels [42]. Other researchers have hypothesised that thrombi may play a direct and significant role in gas exchange abnormalities and multisystem organ dysfunction in COVID-19 pneumonia (https://www.mountsinai.org). Moreover, a few authors have pointed out a presumed role of antiphospholipid antibodies in COVID-19-related thrombotic events [43–45]. So far, it is still controversial whether the presence of these antibodies should be used as evidence for early anticoagulation of patients with COVID-19, knowing that antiphospholipid antibodies are common in the general population especially during infection [46].
Gattinoni and colleagues have highlighted that COVID-19 acute respiratory distress syndrome patients are characterised by the dissociation between relatively well-preserved lung mechanics and severe hypoxemia. These authors have proposed that such severe hypoxemia occurring in compliant lungs could be due to the loss of lung perfusion regulation and hypoxic vasoconstriction [47]. This is reminiscent of the attenuated hypoxic pulmonary vasoconstriction due to pulmonary endothelial cell dysfunction induced by increased mitochondrial oxidative stress in dasatinib-exposed rodents [48]. Last, it has been shown that SARS-CoV-2 elements could be detected in endothelial cells of COVID-19 patients, together with an accumulation of inflammatory cells and evidence of endothelial and inflammatory cells death [49]. This suggests that endotheliitis could be facilitated by SARS-CoV-2 infection and induced in different organs as a direct consequence of viral infection and the host inflammatory response. Consistently, Ackermann et al. found that, beyond diffuse alveolar damage with perivascular T-cell infiltration and widespread thrombosis with microangiopathy, the distinctive vascular features in COVID-19 patients consisted of severe endothelial injury associated with the presence of intracellular virus and disrupted cell membranes, as well as vascular angiogenesis [28].
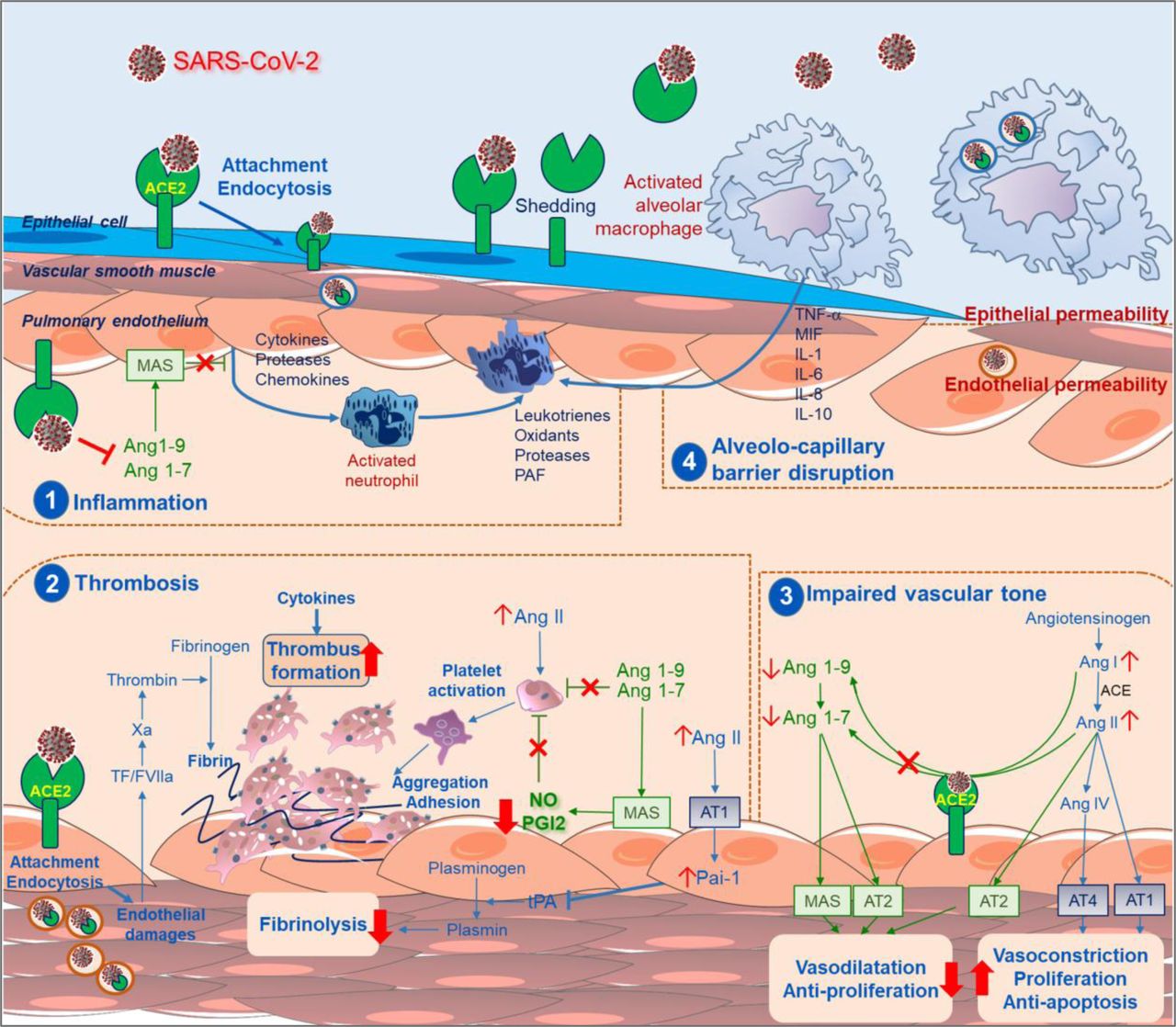
In conclusion, the presence of SARS-CoV-2 virus within the endothelial cells suggests that direct viral effects, as well as perivascular inflammation, may contribute to endothelial cell injury. It is likely that endotheliitis, endothelial injury, endothelial cell dysfunction and impaired microcirculatory function in different vascular beds contributes markedly to life-threatening complications of COVID-19, such as venous thromboembolic disease and multiple organs involvement (fig. 1).
{kind=link}
Schematic representation of hypothetical mechanisms by which the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) causes endothelial dysfunction and pulmonary vascular changes. Following cleavage of its S protein, SARS-CoV-2 was reported to enter human cells via binding to angiotensin-converting enzyme (ACE)2. This transmembrane ACE2 receptor is widely expressed in various pulmonary cells including type II alveolar cells, macrophages, endothelial, smooth muscle cells and perivascular pericytes. This causes uncontrolled inflammation (1), accompanied by micro-thrombosis and occlusion of small pulmonary vessels (2), and impaired endothelial regulation of vascular tone (3), leading to alveolo-capillary barrier disruption (4). Abbreviations and Acronyms : ACE: angiotension converting enzyme; Ang: angiotensin; AT: angiotensin receptor; MAS: macrophage activation syndrome; NO: nitric oxide; PAF: platelet activating factor; PAI-1: plasminogen activator inhibitor-1; PGI2: prostaglandin I2; SARS-CoV-2: Severe Acute Respiratory Syndrome coronavirus 2; TF: tissue factor; tPA: tissue plasminogen activator.
Footnotes
Conflict of interest: Dr. Huertas has nothing to disclose.
Conflict of interest: Dr. MONTANI reports grants and personal fees from Actelion, grants and personal fees from Bayer, personal fees from GSK, personal fees from Pfizer, grants, personal fees and non-financial support from MSD, personal fees from Chiesi, personal fees from Boerhinger, non-financial support from Acceleron, outside the submitted work
Conflict of interest: Dr. Savale reports personal fees and non-financial support from Actelion, personal fees and non-financial support from Bayer, grants and personal fees from GSK, outside the submitted work.
Conflict of interest: Dr. Pichon has nothing to disclose.
Conflict of interest: Dr. TU has nothing to disclose.
Conflict of interest: Dr. Parent reports grants from Bayer, grants from CSL Behring, grants from Sanofi, outside the submitted work.
Conflict of interest: Dr. GUIGNABERT has nothing to disclose.
Conflict of interest: Dr. Humbert reports grants and personal fees from Actelion, grants and personal fees from Bayer, grants and personal fees from GSK, personal fees from Merck, personal fees from United Therapeutics, grants and personal fees from Acceleron, outside the submitted work.
- Received May 6, 2020.
- Accepted June 8, 2020.
- Copyright ©ERS 2020
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










