





Abstract
Respiratory function in Huntington's disease should be monitored from middle stage to preclude respiratory failure http://ow.ly/YXTt300mIQw
To the Editor:
Huntington's disease is an autosomal inherited monogenetic condition in which the mutation is an expansion of the cytosine-adenine-guanine (CAG) repeat sequence at the N-terminal end of the huntingtin gene [1]. More than 40 repeats are associated with neuronal dysfunction and death, predominantly within the striatum resulting in a triad of movement, behaviour and cognitive impairment; other symptoms include weight loss, sleep disturbance and respiratory dysfunction, which may or may not be of primary neurological origin [1–3]. Death occurs 15–30 years after onset of symptoms [1], usually due to pneumonia [4], yet it is not known whether respiratory dysfunction is a feature of late stage disease or whether it appears earlier in the disease evolution. Previous research suggests that dysregulation within the respiratory centre results in irregular breathing patterns [5, 6]; decreased respiratory muscle strength and lung volumes have also been identified [7] which, alongside swallow dysfunction [4], could precipitate respiratory failure. Huntington's disease is a complex long-term condition and contributing factors such as swallow dysfunction, posture, physical inactivity and reduced exercise capacity have not yet been investigated in relation to respiratory function. We conducted a cross-sectional study aiming to characterise respiratory function across all stages of disease and explore primary and secondary contributors to respiratory decline. Given one previous report of respiratory weakness in Huntington's disease, we performed a follow-on study to assess the feasibility of home-based inspiratory muscle training in Huntington's disease.
67 participants testing positive for the Huntington's disease gene were recruited to the cross-sectional study from the South Wales Huntington's disease clinic (Cardiff, UK) between July 2009 and December 2012. Inclusion criteria were: CAG repeat ≥40; aged ≥18 years; able to understand instructions in English; and maintenance of a stable medical regime for 4 weeks prior to the study. Healthy controls (n=39; matched for age, sex, smoking pack-years and fat-free mass) were recruited from relatives, carers and members of Cardiff University. Informed consent was obtained under the Research Ethics Committee for Wales (08/MRE09/65).
Huntington's disease-positive participants were categorised as pre-manifest or manifest based on the presence of motor abnormalities representing an unequivocal diagnosis of Huntington's disease; disease severity was categorised by the Unified Huntington's Disease Rating Scale (total functional capacity scale ranging 0–13) [8]. Outcome measures were: forced vital capacity; forced expiratory volume in 1 s (FEV1)/peak expiratory flow rate (PEFR) ratio; peak cough flow (PCF); maximal inspiratory pressure (MIP); sniff nasal inspiratory pressure (SNIP), maximal expiratory pressure; swallow capacity measured as mL·s-1 during timed swallow of 150 mL of water [9]; metabolic equivalents via the international physical activity questionnaire (short form) [10]; 6-min walk distance (20-m circuit); and thoracic posture via bespoke digital analysis [11].
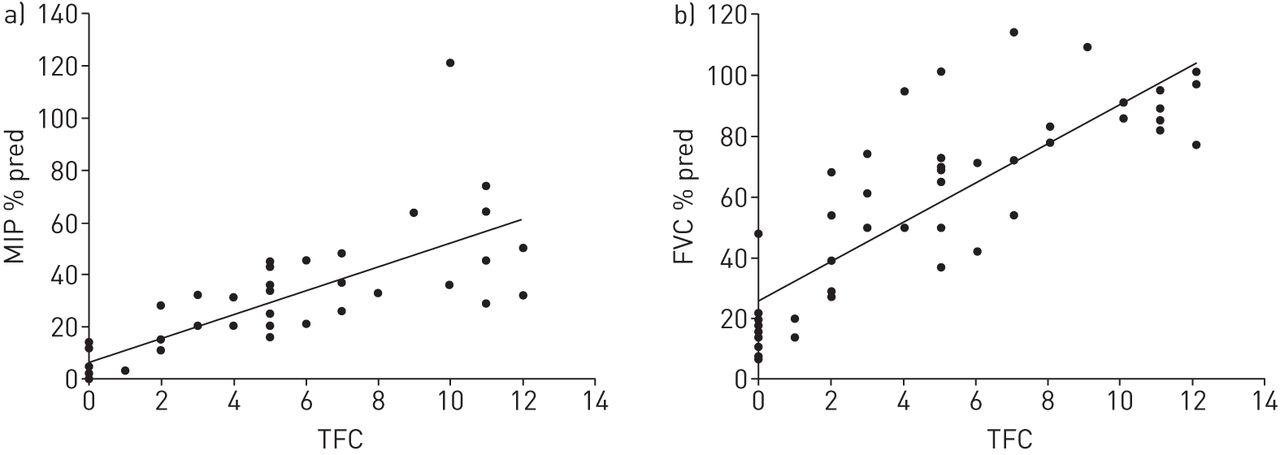
Statistically significant differences were found for all respiratory function variables across healthy, pre-manifest and manifest participants (table 1). Post hoc analyses identified decreased respiratory function in the manifest group compared to healthy controls and pre-manifest groups, with no differences between healthy controls and pre-manifest. Analysis of FEV1/PEFR showed that eight (40%) pre-manifest and 26 (55.3%) manifest participants had a ratio of >8, indicative of central or upper airway obstruction [12]. In manifest participants with Huntington's disease, all measures of respiratory function significantly correlated with disease progression (Spearman's r= 0.716–0.863, p<0.001) (figure 1). Regression analysis indicated PCF <270 L·min−1 [13] when total functional capacity was 5, i.e. middle stage of the disease, suggesting that respiratory intervention may be warranted earlier in the disease process than typically considered.
Comparison of healthy controls, pre-manifest and manifest participants

{kind=link}
Respiratory function and disease progression in people with manifest Huntington's disease. a) Respiratory muscle strength and total functional capacity (TFC). b) Lung volume and TFC. MIP: maximal inspiratory pressure; FVC: forced vital capacity.
Percentage predicted swallow capacity, thoracic angle, physical activity and exercise capacity significantly correlated with respiratory function in manifest participants (Spearman's r= 0.465–0.790, p<0.001). Swallow capacity was normal in all pre-manifest participants and abnormal in 39 (84.8%) manifest participants with Huntington's disease. Physical activity scores were categorised as moderate (median=1502.50 metabolic equivalent minutes·week-1; interquartile range (IQR) 2418.4) for people with pre-manifest Huntington's disease and low (82.50 metabolic equivalent minutes·week-1; IQR 618.80) for people with manifest disease. 6-min walk distance was 78.63% predicted and 27.73% predicted for pre-manifest and manifest participants with Huntington's disease, respectively.
Participants for the follow-on training pilot were selected from the cross-sectional cohort (Ethics: 11/WA/0183; and ISCRTN90741776). Inclusion and exclusion criteria were as for the cross-sectional study, additionally participants were excluded if they were pre-symptomatic and had an MIP >80% pred. 20 participants were randomly allocated to intervention or placebo using a minimisation method [14] to match for age, sex and smoking habit and were blinded to group allocation. The intervention group (n=10) used the POWERbreathe®K3 device (POWERbreathe International Ltd, Southam, UK) for 30 breaths, twice a day at a resistance of 50% of MIP for 6 weeks; resistance was set at 10 cmH2O for the placebo group. The POWERbreathe®K3 device automatically sets resistance based on MIP pressures generated in the first two inspirations. Although resistance based on SNIP would have been more appropriate for the sample, this device provided resistance throughout the range of movement of the inspiratory muscles. A habituation period of 1 week preceded the study. Participants were instructed by the researcher who provided support through alternate weekly phone calls and home visits. The primary outcome measure was SNIP and the secondary outcome was cough efficacy as measured by PCF, the measurement was not blinded. Adherence was measured as number of training sessions as recorded by the POWERbreathe®K3 device.
All participants had MIP and SNIP <80% pred. Adherence to inspiratory muscle training was similar for both intervention and placebo groups: 70.67±26.35% and 74.53±21.03%, respectively. For those that completed the study, SNIP increased from 47.60±33.96 cmH2O to 53.60±28.31 cmH2O (intervention: n=5) and from 46.00±20.06 cmH2O to 53.00±18.17 cmH2O (placebo: n=7) and PCF increased from 415.80±153.57 L·min−1 to 448±144.97 L·min−1 (intervention) and from 334.00±101.64 L·min−1 to 370.71±89.00 L·min−1 (placebo). With nonsignificant increases in SNIP and PCF within both groups, further analyses were conducted pooling the groups to explore whether participating in the programme, irrespective of resistance, changed respiratory function. Post-intervention mean difference for SNIP was 8.07 cmH2O (effect size 0.36) and PCF 49.39 L·min−1 (effect size 0.37). The small effect sizes suggest that regular breathing exercises, irrespective of added resistance, could improve cough efficacy and respiratory muscle strength.
We present data that gives a clear indication that respiratory failure in Huntington's disease is not simply a consequence of a neurodegenerative condition. The decline in respiratory function is integral to the disease process and we suggest that regular breathing exercises may increase the capacity of the respiratory muscles and improve cough effectiveness. This study expands on previous research findings of reduced lung volume and respiratory muscle strength in Huntington's disease [7] by identifying that: upper airway changes occur in pre-manifest Huntington's disease; cough effectiveness is reduced in the mid stage of the disease; and posture, physical activity and exercise capacity influence respiratory function in Huntington's disease. Our data provides a holistic representation of respiratory function in Huntington's disease across the disease life cycle. We present comprehensive data of physiological variables of decreased lung volume and respiratory muscle strength and upper airway obstruction that are responsible for respiratory failure in people with Huntington's disease and suggest that clinicians monitor respiratory function from at least the middle stage of the disease. Further research is needed to identify the appropriate interventional strategies based on confirmation of feasibility and effectiveness of regular breathing exercises in this study.
Footnotes
Clinical trials: This study is registered at ISRCTN with the identifiers ISRCTN90741776 and ISRCTN72770961
Support statement: Funding was received from the European Huntington's Disease Network, Physiotherapy Research Foundation UK and Research Capacity Building Collaboration Wales. Funding information has been deposited with Open Funder Registry.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received December 13, 2015.
- Accepted April 28, 2016.
- Copyright ©ERS 2016
ERJ Open articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.










