





Abstract
Background The effects of convalescent plasma (CP) therapy in hospitalised patients with coronavirus disease 2019 (COVID-19) remain uncertain. This study investigates the effect of CP on clinical improvement in these patients.
Methods This is an investigator-initiated, randomised, parallel arm, open-label, superiority clinical trial. Patients were randomly (1:1) assigned to two infusions of CP plus standard of care (SOC) or SOC alone. The primary outcome was the proportion of patients with clinical improvement 28 days after enrolment.
Results A total of 160 (80 in each arm) patients (66.3% critically ill, 33.7% severely ill) completed the trial. The median (interquartile range (IQR)) age was 60.5 (48–68) years; 58.1% were male and the median (IQR) time from symptom onset to randomisation was 10 (8–12) days. Neutralising antibody titres >1:80 were present in 133 (83.1%) patients at baseline. The proportion of patients with clinical improvement on day 28 was 61.3% in the CP+SOC group and 65.0% in the SOC group (difference −3.7%, 95% CI −18.8–11.3%). The results were similar in the severe and critically ill subgroups. There was no significant difference between CP+SOC and SOC groups in pre-specified secondary outcomes, including 28-day mortality, days alive and free of respiratory support and duration of invasive ventilatory support. Inflammatory and other laboratory marker values on days 3, 7 and 14 were similar between groups.
Conclusions CP+SOC did not result in a higher proportion of clinical improvement on day 28 in hospitalised patients with COVID-19 compared to SOC alone.
Abstract
In this open-label, randomised clinical trial, two infusions of convalescent plasma therapy plus standard of care compared to standard of care did not result in higher proportion of clinical improvement on day 28 in patients with severe COVID-19 https://bit.ly/2TXuB6S
Introduction
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), can cause severe illness in a considerable proportion of infected patients leading to severe progressive pneumonia, multiple organ dysfunction and death [1, 2].
Passive immunotherapy using convalescent plasma (CP) collected from COVID-19 recovered patients has been advocated for the treatment of severe cases of this disease [3]. The United States Food and Drug Administration issued an emergency-use authorisation for CP for the treatment of hospitalised patients with COVID-19 based on results of observational studies showing that CP was safe and could be associated with better clinical outcomes [4, 5]. Nevertheless, the two existing randomised clinical trials at the time of authorisation [6, 7] and further multicentre randomised clinical trials [8, 9] have failed to demonstrate significant clinical benefit of CP in patients with severe COVID-19. The long duration of disease when intervention occurred and low neutralising antibody titres in administered plasma may, at least partially, explain the absence of significant improvement in clinical outcomes in intervention groups in two of these trials [6, 7]. The other two (larger) clinical trials did not find any benefit of CP on clinical outcomes. However, these studies used anti-SARS-CoV-2 spike IgG as a surrogate for neutralising antibodies titres, impairing inferences that could be made on the baseline patient status regarding these antibodies and the investigated intervention [8, 9].
Given the heterogeneity regarding CP characteristics, including volume, number of doses and neutralising antibody titres, as well as distinct levels of pre-existing antibody titres at baseline in both intervention and control groups, further clinical trials with different administration strategies and distinct populations are necessary to better define the role of this therapy in hospitalised patients with severe COVID-19. In the present randomised clinical trial, we assessed the effect of two doses of 300 mL of CP therapy administered in the first 14 days of symptoms onset on clinical improvement in severe and critically ill COVID-19 patients.
Methods
Study design and oversight
PLACOVID was an investigator-initiated, unicentric, randomised, parallel-arm, open-label, superiority clinical trial performed at a single COVID-19 reference hospital in Porto Alegre, Brazil.
This study was approved by the Brazilian National Commission for Research Ethics and the institutional review board of Hospital de Clínicas de Porto Alegre (approval number, 20-0158). Written informed consent was obtained from all study participants or their legal representatives. The trial was overseen by an external and independent data and safety monitoring board. The trial protocol and statistical analysis plan are available in supplementary material 1. The trial was registered with the number NCT04547660 (https://clinicaltrials.gov/ct2/show/NCT04547660).
Participants
Patients admitted to the hospital were assessed for eligibility if they were aged ≥18 years, had a positive reverse transcriptase (RT)-PCR for SARS-CoV-2 (supplementary material 2), had <15 days since initial symptom onset and had severe respiratory disease, as defined by the presence of at least one of the following: respiratory rate >30 breaths·min-1 in room air; peripheral oxygen saturation (SpO2) ≤93% in room air; arterial partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ≤300; need for supplemental oxygen to maintain SpO2 >95%; need for supplemental oxygen by high-flow nasal cannula, noninvasive ventilation or invasive mechanical ventilation. Exclusion criteria were impossibility for any reason to perform the first plasma infusion within 14 days of the onset of symptoms; use of immunosuppressive drugs for other non-COVID-19 underlying diseases in the 30 days before enrolment; pregnancy; history of serious adverse reactions such as transfusion anaphylaxis; disagreement of attending physician; and participation in other interventional randomised clinical trials.
Plasma donation procedures
A full description of plasma donation selection and procedures is shown in supplementary material 2.
Randomisation and interventions
Patients were randomly assigned in a 1:1 ratio to receive two infusions 48 h apart of 300 mL of CP plus standard of care (SOC) or SOC alone. Randomisation was computer-generated with random block sizes of two or four and stratified according to the unit of hospitalisation on enrolment (medical ward or intensive care unit (ICU); unit of hospitalisation on enrolment was used as a proxy for disease severity). Patients and investigators were unblinded, except interviewers performing follow-up telephone calls, who were unaware of the assigned trial group.
The SOC for COVID-19 was at the discretion of the treating physicians. The use of glucocorticoids, other immunomodulators, antibiotic agents and antiviral agents was allowed. Remdesivir was not available in Brazil during the trial period.
Clinical and laboratory data
Definitions of baseline variables assessed at baseline are presented in the supplementary methods (supplementary material 1). Neutralising antibodies were determined in all donor plasma units and in patient serum collected on days 0 and 3 (after the second plasma infusion) after enrolment, following a previously described protocol [10]. Nasal and oropharyngeal swabs were collected at day 7 after enrolment or at hospital discharge. Blood samples were collected on days 0 (pre-infusion), 3 (post-second infusion), 7 and 14 after enrolment in hospitalised patients.
Outcomes and follow-up
The primary outcome was the proportion of patients with clinical improvement 28 days after enrolment. Clinical improvement was defined as hospital discharge or reduction of 2 points in a six-level ordinal scale. Levels on the scale were defined as follows. 1=not hospitalised; 2=hospitalised and not receiving supplemental oxygen; 3=hospitalised and receiving supplemental oxygen; 4=hospitalised and receiving oxygen supplementation administered by a high-flow nasal cannula or noninvasive ventilation; 5=hospitalised and receiving mechanical ventilation or extracorporeal membrane oxygenation; and 6=death. Pre-specified secondary outcomes included RT-PCR for SARS-CoV-2 from nasal and oropharyngeal swab at day 7 from enrolment or hospital discharge (if earlier than 7 days); clinical status assessed using the six-level ordinal scale and all-cause mortality at days 14 and 28 after enrolment; time to hospital discharge and days alive and free of supplemental oxygen support (nonsurvivors and patients requiring oxygen support at day 28 were assigned as 0 supplemental oxygen support-free days) within 28 days of enrolment; Sequential Organ Failure Assessment (SOFA) score and National Early Warning Score (NEWS) 2 on day 7 after enrolment; and length of invasive ventilatory support (for those who received mechanical ventilation). Adverse events were assessed using the Common Terminology Criteria for Adverse Events (CTCAE) grade 3 or 4 up to day 28 after enrolment or hospital discharge. Other pre-specified exploratory outcomes were levels of serum inflammatory markers and cytokines, measured on days 3, 7 and 14 after enrolment (supplementary material 2).
Patients were followed daily up to day 28 after enrolment or hospital discharge by researchers who were aware of the trial-group assignments. For patients who were transferred to another hospital before day 28, a structured telephone call to the patient or the patient's family was conducted by an interviewer to assess the level on the ordinal scale at day 28.
Sample-size calculation and protocol changes
We had originally planned for the trial to include 160 patients considering all-cause mortality within 28 days as the primary outcome and an absolute difference between arms of 20% to achieve a power of 80%, using the formula for two binomial proportions and two-sided tests, as described by Rosner [11]. However, due to the evolving knowledge on COVID-19, the steering committee assumed that a reduction of 20% in mortality would be very unlikely to occur and that estimated proportions for survival and death within 28 days were better suited for clinical response. Therefore, it was decided to submit a protocol amendment on 27 July 2020 (when eight patients had been included in the trial) modifying the primary outcome to clinical improvement on day 28 after enrolment.
In the revised sample-size calculation, assuming a proportion of clinical improvement of 60% in the SOC group (supplementary material 2), a sample of 160 patients (80 in each arm) was estimated to achieve a power of 80% to detect an absolute difference of ≥20% in the proportion of patients with clinical improvement at day 28 with a two-sided α-level of 0.05. Other modifications are detailed in the study protocol in supplementary material 1.
Statistical analysis
Data were primarily analysed according to the intention-to-treat principle. The proportion of patients with clinical improvement on day 28 and relative risk were assessed using robust Poisson regression. Pre-specified subgroups were defined according to the unit of hospitalisation (medical ward (considered severe patients) or ICU (considered critically ill patients)) and mechanical ventilation needed on enrolment. Consistency of intervention effects on the primary outcome across these subgroups was assessed by means of interaction tests.
A post hoc sensitivity analysis was performed for the primary outcome considering a per-protocol population. Secondary outcomes were compared generalised linear models, according to the probability distribution of the outcome, or using the Wilcoxon–Mann–Whitney, test as appropriate. The potential effect of variables with a p-value ≤0.20 at the baseline on the primary outcome was addressed in Poisson regression models. An additional exploratory analysis considered the clinical improvement outcome as a reduction of 1 point in the ordinal scale.
One pre-planned interim analysis for efficacy and safety evaluation after 80 patients with complete follow-up was conducted (supplementary material 2). The stopping rule for efficacy and safety was a p-value<0.05. There was no adjustment in the final threshold for statistical significance for sequential analysis.
All analyses were performed using the R software version 4.0.2 (www.r-project.org). No adjustments for multiplicity were performed. Thus, the results of secondary outcomes and subgroup analyses should be interpreted as exploratory. A two-sided p-value of <0.05 was considered statistically significant.
Results
Patients
From 15 July to 10 December 2020, 443 patients were assessed for eligibility. Of these, 160 were eventually enrolled: 80 in the CP+SOC group, and 80 in the SOC alone group (figure 1). The follow-up was completed on 7 January 2021. A total of 106 (66.3%) patients were located in the ICU and 54 (33.7%) were in the medical ward at randomisation. Baseline characteristics are shown in table 1. The median (interquartile range (IQR)) age was 60.5 (48.0–68.0) years, 93 (58.1%) were male and the median (IQR) time from symptom onset to randomisation was 10 (8–12) days. 133 (83.1%) patients presented neutralising antibody titres above 1:80 at randomisation (median 1:1280, IQR 1:320–1:2560). All but two (1.2%) patients were receiving glucocorticoids at the time of entry into the trial. The baseline characteristics of the participants enrolled in CP+SOC group and of those enrolled in the SOC alone group were similar, except for median neutralising antibody titres, which were significantly higher in the control than in the intervention group, and interleukin-6 levels, which were significantly higher in the intervention group than in the control group (table 1 and supplementary material 2).
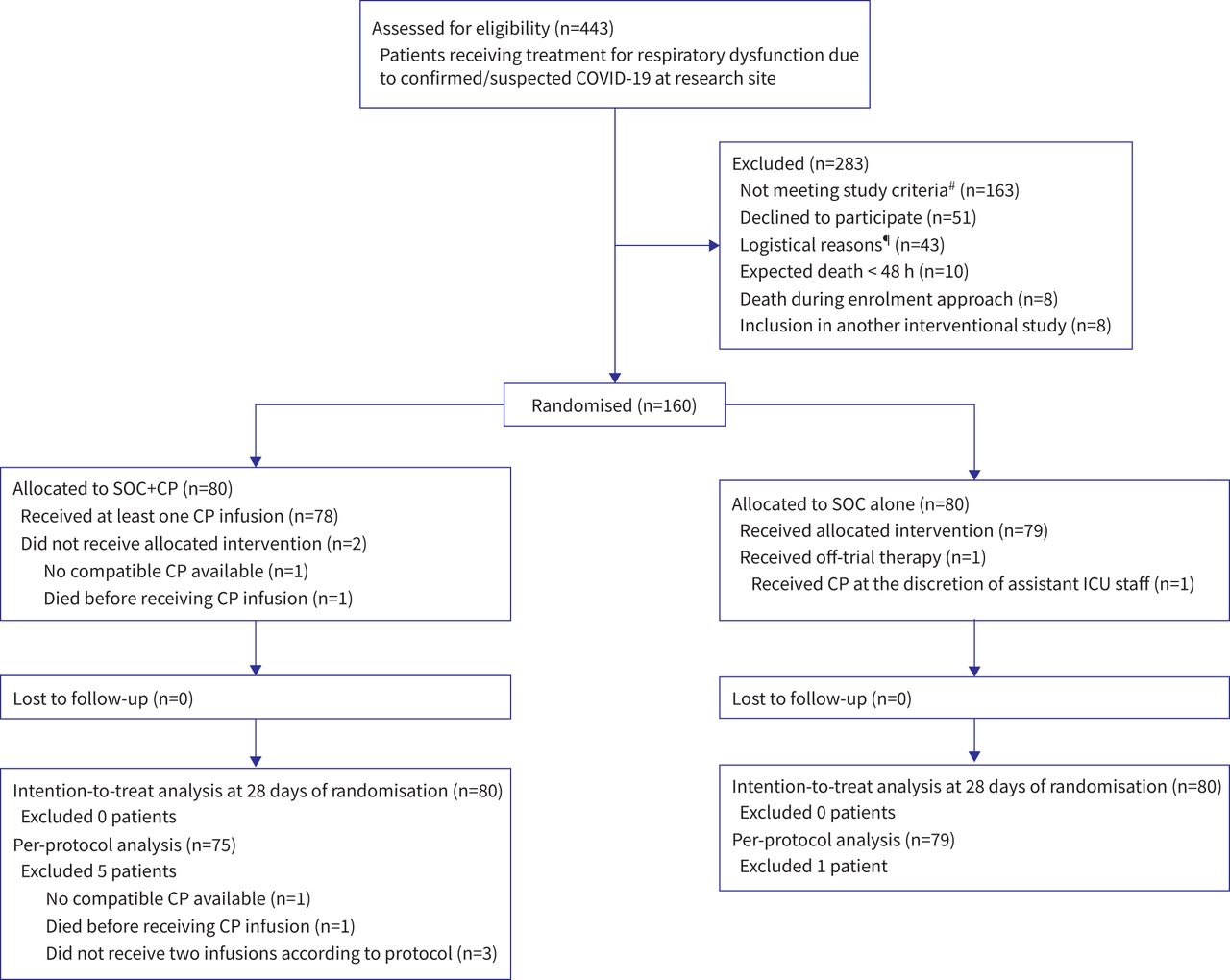
Flow diagram of patients in the clinical trial. COVID-19: coronavirus disease 2019; SOC: standard of care; CP: convalescent plasma; ICU: intensive care unit. #: not meeting inclusion criteria: >14 days of symptoms (n=88); negative severe acute respiratory syndrome coronavirus 2 reverse transcriptase PCR (n=38); previous use of immunosuppressants (n=30); no need for oxygen support (n=5); age <18 years (n=2). ¶: screened patients were sequentially approached until a maximum of four subjects were enrolled daily due to limited capacity from the research team to collect blood samples and infuse convalescent plasma within advocated time interval. Eligible patients exceeding this limit were approached the next day or were excluded from the study if no longer compliant with inclusion criteria.
Baseline characteristics of patients
Interventions
62 (77.5%) patients received CP from the same donor at each infusion, while 15 (18.8%) received the second infusion from a different donor. The median (IQR) neutralising antibody titres from donor plasma administered to patients from the intervention group was 1:320 (1:160–1:960), which was significantly lower than baseline neutralising antibody titres of patients prior to the infusion (p<0.001). Only five donors’ plasma had neutralising antibody titres lower than 1:80 (four 1:40 and one 1:20). Other characteristics of CP donors are shown in supplementary material 2.
Two (1.3%) patients allocated to CP did not receive any intervention (one due to the lack of compatible plasma units; one patient died before receiving transfusion) and another two (1.3%) patients did not receive the second plasma infusion. One patient allocated to CP received four additional plasma infusions at the discretion of the attending physician. One patient allocated to the control group received one unit of CP, at the discretion of the ICU team.
On day 3, there was a significantly higher increase in neutralising antibody titres in the intervention than in control group (p=0.001) in relation to titres at randomisation (day 0) (figure 2). The median (IQR) neutralising antibody neutralising titres on day 3 was not significantly different between CP and SOC groups (1:5120, 1:2560–1:10 240 versus 1:2560, 1:1920–5120; p=0.19) (figure 2).

Distribution of neutralising antibody titres in convalescent plasma and standard-of-care (control) groups at randomisation and on day 3. Each colour indicates the proportion of patients with a given neutralising antibody titre. Titres of 1:10 or 1:20 were grouped in the 1:20 category. At randomisation, n=80 (convalescent plasma) and n=78 (control); on day 3, n=78 (convalescent plasma) and n=76 (control).

{kind=link}
{kind=link}
{kind=link}
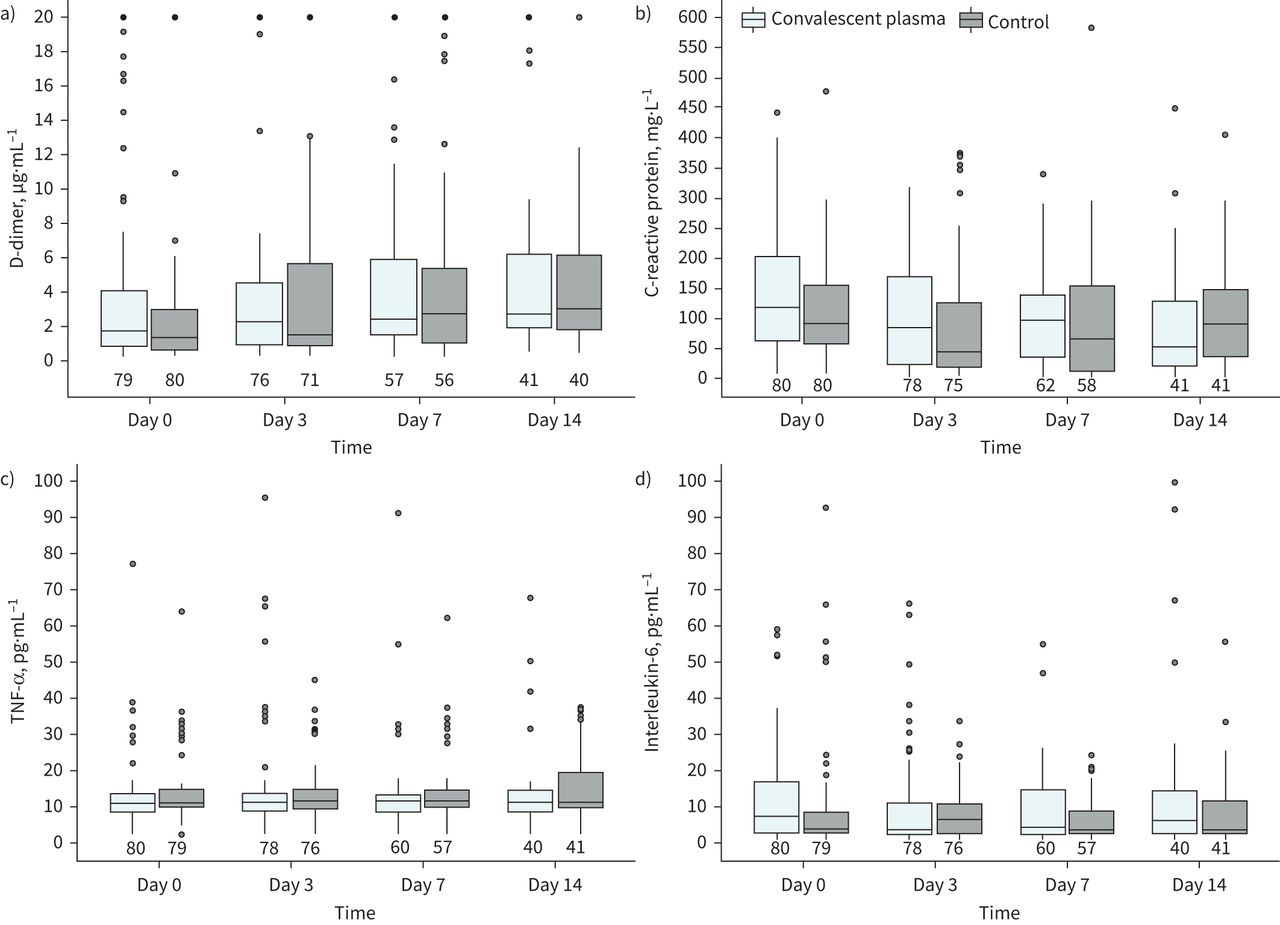
Inflammatory markers at randomisation, and on days 3, 7 and 14. a) D-dimer; b) C-reactive protein; c) tumour necrosis factor (TNF)-α; d) interleukin-6. The box plot inner horizontal lines indicate median; boxes, interquartile range (25th and 75th percentiles); whiskers extend to the most extreme observed values with 1.5 times the interquartile range of the nearer quartile, and dots represent observed values outside that range. The numbers of patients evaluated at each time point in both convalescent plasma and control groups are at the bottom of the figure.
Primary outcome
On day 28, there was no significant difference between the CP+SOC group and the SOC alone group in the proportion of patients with clinical improvement (61.3% versus 65.0%; difference −3.7%, 95% CI −18.8–11.3; relative risk 0.94, 95% CI 0.74–1.19; p=0.623) (table 2). Results for the per-protocol population were similar to those of the main analysis (supplementary material 2). In subgroup analyses, tests for interaction were not statistically significant for subgroups defined by the unit of admission, need of mechanical ventilation, age and neutralising antibody titres at baseline (supplementary material 2).
Primary and secondary end-points
Secondary outcomes
CP+SOC group effects were not significantly different from SOC alone group for 28-day mortality (22.5% versus 16.3%; difference 6.2%, 95% CI −7.5–20.7%; relative risk 1.38, 95% CI 0.73–2.63; p=0.32), proportion of scores on the six-level ordinal scale on day 28 (p=0.64) and median of days alive and free of respiratory support within 28 days (11.0 versus 7.5; difference −0.6 days, 95% CI −3.9–2.6; p=0.44). There was no significant difference between groups in other secondary outcomes (table 2; supplementary material 2).
The proportion of patients with a positive RT-PCR for SARS-CoV-2 from nasal and oropharyngeal swab on day 7, or on discharge if earlier, was similar in the CP+SOC and SOC alone groups (76.3% versus 74.1%; difference 2.2%, 95% CI −13.6–17.9%; relative risk 1.03, 95% CI 0.84–1.27; p=0.79) (table 2). There was no statistically significant difference in inflammatory markers and other laboratory parameters between groups on days 3, 7 and 14 in both the all-patients population and in those who had completed the three-collection sequence (figure 3, supplementary material 2).
Adverse events
The safety population included 79 patients who received at least one infusion of CP and 81 patients who received only SOC. 52 (65.8%) and 48 (59.3%) patients presented an adverse effect in CP+SOC and SOC-alone groups, respectively (absolute difference 5.0%, 95% CI −10.0–20.1%; relative risk 1.08, 95% CI 0.85–1.38; p=0.51). CTCAE grade 3 or 4 adverse effects were noted in 50 (63.3%) and 44 (54.3%) patients in the intervention and control groups, respectively (absolute difference 7.5%, 95% CI −7.8–22.8%; relative risk 1.14, 95% CI 0.88–1.48; p=0.34). A full description of adverse effects is shown in supplementary material 2.
Post hoc analyses
There was no significant difference in no pre-specified subgroup analysis by age and neutralising antibody titres at baseline (supplementary material 2). There was no significantly difference between intervention and SOC groups in Poisson regression models including variables with a p-value ≤0.20 at the baseline (table 3). Furthermore, there was no significant difference in clinical improvement on day 28, i.e. a 1-point reduction in the ordinal scale (61.3% versus 68.8% in the intervention and control groups, respectively; relative risk 0.89, 95% CI 0.71–1.12; p=0.33).
Sensitivity analysis by Poisson regression models# including variables with a p-value ≤0.20 at baseline
Discussion
In this randomised clinical trial with severe and critically ill COVID-19 patients, CP therapy administered in the first 14 days of the onset of symptoms plus SOC did not increase the proportion of clinical improvement on day 28 compared with SOC alone. Similar results were found in both critically ill and patients hospitalised at medical ward subgroups. These findings are consistent with previous randomised clinical trials that could not find significant benefit of CP in hospitalised patients with COVID-19 [6–9].
There were also no significant differences in clinical and laboratory outcomes between the intervention and control groups, including 14- and 28-day mortality, clinical status on days 14 and 28 assessed by an ordinal scale, days free of ventilation, days of hospitalisation and SOFA and NEWS2 scores. No difference was observed considering a 1-point reduction in the ordinal scale as clinical improvement.
One strength of our study is that virtually all patients were treated with corticosteroids, mostly dexamethasone, as SOC and other drugs were not used. Additionally, this study was the first to evaluate some laboratory exams in patients’ follow-up. The demonstration of absence of difference in these markers are consistent with clinical findings and help to reduce the level of uncertainty on the potential benefit of CP in severe COVID-19. Notably, in contrast to Li et al. [6] we could not find any difference in SARS-CoV-2 RT-PCR positivity rate between groups.
As found in a previous trial [7], most of the patients included in the study have already presented high levels (above 1:80) of neutralising antibody titres at randomisation. These titres were even higher in the SOC group (>75% of patients with titres equal to or greater than 1:640). Two infusions of 300 mL of CP increased the levels of these antibodies on day 3 in the intervention group. The increase in neutralising antibody titres from randomisation to day 3 was significantly higher in intervention group, and although the levels were higher in intervention than in control on day 3, this difference was not statistically significant probably because baseline levels in the former group was lower than the latter. Nonetheless, this increase seemed to have no impact on both clinical and laboratory outcomes, as indicated by the absence of any significant difference of inflammatory markers between groups in any point of collection from day 0 to 14. It must be acknowledged that the presence of high levels of neutralising antibodies titres at randomisation favours the null hypothesis, even though the effect on primary outcome was not affected when adjusted for this variable in the Poisson regression model. Furthermore, it is highly relevant from a pragmatic perspective, i.e. increment in antibody response in patients through passive administrations does not seem to be worthy in patients with severe COVID-19.
Notably, patients in the intervention group presented significantly higher levels of interleukin-6 at randomisation. Although interleukin-6 levels did not significantly modify the effect of convalescent plasma on the outcome, like other variables analysed in Poisson regression models, and at first we attributed this as a casual difference that may be observed even with the randomisation process, we cannot fully rule out that patients in intervention arm might be more severely ill. However, as shown in sensitivity analysis, if present, this imbalance did not affect the main results of the study.
A recently published meta-analysis evaluating the effect of CP on mortality and other clinical outcomes, including pre-print publications and a press release of one randomised trial, could not find any significant difference of this strategy from SOC or placebo [12]. Given the heterogeneity of doses, neutralising antibody titres and time of CP administration, along with the fact that most randomised trials have been interrupted prematurely, as pointed out by the authors, the certainty of the evidence was low to moderate for all-cause mortality and low for other clinical outcomes. We updated that meta-analysis using the same methodology, including data previously available only as a press release [9], and the PLACOVID trial for mortality. The updated result remains nonsignificant, with low inconsistency and narrow confidence intervals (relative risk 0.98, 95% CI 0.81–1.19; p=0.29; I2=16%) (supplementary material 2).
This study has some limitations. First, it is an open-label study and data collectors were not blinded to the patients’ group assignment. Despite not finding a positive effect of intervention, potential biases associated with this design cannot be ruled out completely. Second, our clinical trial is a single-centre study in a COVID-19 reference tertiary-care university-affiliated hospital, which may impair the generalisability of the findings; however, the overall findings point towards the same direction as previous multicentre studies. Third, this study is composed mostly of critically ill patients, a group of patients in whom less potential benefit could be expected. Nonetheless, similar results were found in both critically ill and patients hospitalised in the medical ward. Finally, we were underpowered to evaluate efficacy in patients with low neutralising antibody titres. Despite the low number of patients, the exploratory analysis of patients with titre less than 1:160 indicates a change in the direction of the effect (supplementary material 2). Along with previous studies suggesting a potential benefit with CP [13] or monoclonal antibodies in early periods of mild-to-moderate COVID-19 [14, 15], patients with severe COVID-19 and low levels of neutralising antibodies might still be a group of interest for future studies with passive immunotherapy.
In conclusion, in severe or critically ill COVID-19 patients, almost all of whom were receiving corticosteroids as SOC, CP+SOC did not result in a higher proportion of clinical improvement on day 28 compared to SOC alone.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 1 ERJ-01471-2021.Supplement_1
Supplementary material 2 ERJ-01471-2021.Supplement_2
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01471-2021.Shareable
Acknowledgements
We are grateful to Patrícia Prolla and the “Grupo de Pesquisa e Pós-Graduação (GPPG)” team from Hospital de Clínicas de Porto Alegre (Porto Alegre, Brazil) for their significant administrative support during the study implementation and execution.
Footnotes
The PLACOVID Study Group at Hospital de Clínicas de Porto Alegre are: Alecsandra Formentin Bello, Alldren da Silva de Souza, Ana Claudia Tonelli de Oliveira, Andréia R. Malaquias, Ane Katiussa Siqueira Fröhlich, Anelise Bergmann Araújo, Antônia Cícera da Silva Araújo, Bruna Blos, Carolina Rodrigues Cohen, Cristiane Tavares Borges, Cristiano Rossa da Rocha, Daniela Michelim Rodrigues Speranza, Delany da Silva Oliveira, Dimitris Rucks Varvaki Rados, Francine Bonacina, Gabrielle Dias Salton, Giovana Zucchetti, Isabel Cristina Freitas, Juliana Goncalves Constante, Juliana Monteiro Furlan, Karine Kleber, Laís Pelentier Vieira, Leonardo Martins Pires, Liane Marise Rohsig, Luciana do Nascimento Vargas, Marize S.V. Leão, Melissa Helena Angeli, Patrícia Paim Ferreira Seltenreich, Patrícia Santos da Silva, Rafael Selbach Scheffel, Renata Eliane Boehm, Renato G.B. de Mello and Thabyta Silva Franco de Souza.
This article has supplementary material available from erj.ersjournals.com
This article has an editorial commentary: https://doi.org/10.1183/13993003.02076-2021
This study is registered at Clinicaltrials.gov as NCT04547660. The authors encourage interested parties to contact the corresponding author with data-sharing requests, including for access to additional unpublished data.
Conflict of interest: L. Sekine has nothing to disclose.
Conflict of interest: B. Arns has nothing to disclose.
Conflict of interest: B.R. Fabro has nothing to disclose.
Conflict of interest: M.M. Cipolatt has nothing to disclose.
Conflict of interest: R.R.G. Machado received support from “Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)” (2017/24769-2).
Conflict of interest: E.L. Durigon has nothing to disclose.
Conflict of interest: E. Parolo has nothing to disclose.
Conflict of interest: J.A.S. Pellegrini has nothing to disclose.
Conflict of interest: M.V. Viana has nothing to disclose.
Conflict of interest: P. Schwarz has nothing to disclose.
Conflict of interest: T.C. Lisboa has nothing to disclose.
Conflict of interest: J.M.S. Dora has nothing to disclose.
Conflict of interest: J.P. Portich has nothing to disclose. A.A. Paz has nothing to disclose. L. Silla has nothing to disclose. A.M. Balsan has nothing to disclose.
Conflict of interest: F.d-S. Schirmer has nothing to disclose.
Conflict of interest: J.P.M. Franz has nothing to disclose.
Conflict of interest: L.M. da-Silveira has nothing to disclose.
Conflict of interest: R.C. Breunig has nothing to disclose.
Conflict of interest: V. Petersen has nothing to disclose.
Conflict of interest: M. Sosnoski has nothing to disclose.
Conflict of interest: N.F. Mesquita has nothing to disclose.
Conflict of interest: F.C.Z. Volpato has nothing to disclose.
Conflict of interest: D. Sganzerla has nothing to disclose.
Conflict of interest: M. Falavigna has nothing to disclose.
Conflict of interest: R.G. Rosa received research grants from Brazilian Ministry of Health.
Conflict of interest: A.P. Zavascki is a research fellow of the National Council for Scientific and Technological Development (CNPq), Ministry of Science and Technology, Brazil (304226/2018-1), and receives a research grant not related to this work from Pfizer (WI242215 2018).
Support statement: The study was funded by “Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul” (FAPERGS) (grant 16/2551-0000242-8), “Fundação de Amparo à Pesquisa do Estado de São Paulo” (FAPESP) (grants 2020/06409-1 and 2016/20045-7) and “Instituto Cultural Floresta”. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 30, 2021.
- Accepted June 16, 2021.
- Copyright ©The authors 2022.
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org










