





Abstract
Respiratory syncytial virus (RSV) is the major cause of acute bronchiolitis in infants under 2 years old. Necroptosis has been implicated in the outcomes of respiratory virus infections. We report that RSV infection triggers necroptosis in primary mouse macrophages and human monocytes in a RIPK1-, RIPK3- and MLKL-dependent manner. Moreover, necroptosis pathways are harmful to RSV clearance from alveolar macrophages. Additionally, Ripk3−/− mice were protected from RSV-induced weight loss and presented with reduced viral loads in the lungs.
Alveolar macrophage depletion also protected mice from weight loss and decreased lung RSV virus load. Importantly, alveolar macrophage depletion abolished the upregulation of Ripk3 and Mlkl gene expression induced by RSV infection in the lung tissue.
Autocrine tumor necrosis factor (TNF)-mediated RSV-triggered macrophage necroptosis and necroptosis pathways were also involved in TNF secretion even when macrophages were committed to cell death, which can worsen lung injury during RSV infection. In line, Tnfr1−/− mice had a marked decrease in Ripk3 and Mlkl gene expression and a sharp reduction in the numbers of necrotic alveolar macrophages in the lungs. Finally, we provide evidence that elevated nasal levels of TNF are associated with disease severity in infants with RSV bronchiolitis.
We propose that targeting TNF and/or the necroptotic machinery may be valuable therapeutic approaches to reduce the respiratory morbidity caused by RSV infection in young children.
Abstract
RIPK1-, RIPK3- and MLKL-dependent alveolar macrophage necroptosis triggered by RSV infection is mediated by autocrine TNF and harmful to viral clearance. Alveolar macrophage necroptosis may drive RSV disease pathogenesis. https://bit.ly/3fS34uw
Introduction
Respiratory syncytial virus (RSV) is the most frequent cause of hospitalisation due to acute bronchiolitis and viral pneumonia in infants under 2 years of age [1, 2] and has been estimated to be responsible for up to 200 000 deaths every year worldwide [2, 3]. Infants born preterm or with high-risk medical conditions, such as congenital heart disease or chronic lung disease, have an increased risk of developing severe RSV disease [4]. Accumulating evidence suggests that RSV infection in childhood may predispose to recurrent wheezing and asthma later in life [5, 6]. Although the World Health Organization has designated RSV as a high-priority target for vaccine development, so far there is no clinically approved RSV vaccine. Therefore, understanding how RSV interacts with the host is of the utmost importance to better characterise disease pathogenesis and to develop new therapies.
Alveolar macrophages (AMs) account for nearly 95% of leukocytes in the airways and are the first immune cells to encounter pulmonary pathogens [7, 8]. For this reason, their response to infection can critically influence the outcomes of infectious diseases [7, 9]. In this scenario, emerging evidence has revealed that AM death plays an important role in determining the pathogenesis of pulmonary viral infections [10, 11].
Necroptosis is considered a pro-inflammatory form of cell death that leads to the release of intracellular contents, which may act as damage-associated molecular patterns (DAMPs), stimulating immune cells [12]. Necroptosis is driven by the assembly of a multi-protein complex consisting of receptor-interacting protein kinase 1 (RIPK1) and RIPK3, leading to the activation of the pseudokinase mixed-lineage kinase domain-like (MLKL) [13, 14]. Necroptotic cell death is effective in restraining the spread of intracellular pathogens, such as viruses, by eliminating replication sites [15, 16]; however, it imposes a cost on the host by exacerbating inflammation and tissue injury following infection. Accordingly, it has been shown that influenza A H1N1 virus induces necroptosis in alveolar epithelial cells and that necroptosis is a pathological feature of the infection, being responsible for the high morbidity and mortality of infected animals [17]. The lack of RIPK3 causes decreased cytokine expression and alleviates influenza A H7N9 virus-induced acute lung injury in mice [18]. It has been recently reported that RSV triggers alveolar epithelial cells and THP-1 cell necroptosis [19, 20]. Nevertheless, whether RSV induces AM necroptosis and whether its effect is protective or detrimental to the host are yet to be demonstrated.
Here, we report that an active RSV infection is necessary and sufficient to trigger necroptosis in primary mouse macrophages and human monocytes in a RIPK1-, RIPK3- and MLKL-dependent manner. Moreover, the activation of necroptosis pathways is detrimental to viral clearance. Ripk3−/− mice were protected from RSV-induced weight loss and presented with reduced viral loads in the lungs. Surprisingly, AMs have been shown to play a harmful role in RSV infection, and AM depletion abolished the upregulation of Ripk3 and Mlkl mRNA levels in the lung tissue induced by RSV infection. Moreover, autocrine tumor necrosis factor (TNF) mediates RSV-triggered macrophage necroptosis and Tnfr1−/− mice had a marked decrease in the expression of Ripk3 and Mlkl mRNA and a sharp reduction in the number of dead AMs in the lungs. These data suggest that TNF-mediated AM necroptosis plays a significant role in the pathogenesis of RSV infection.
Methods
Virus preparation
Virus production of RSV A2 strain (kindly donated by Fernando Polack, Fundación Infant, Argentina) was obtained in VERO cells (ATCC CCL-81) cultured in Opti-MEM medium (Gibco by Life Technologies, Grand Island, NY, USA) with 2% fetal bovine serum (FBS) (Gibco by Life Technologies) at 37°C under 5% CO2. To assess viral titre, VERO cells were infected with RSV in medium without serum followed by a carboxymethylcellulose plaque assay. Lysis plate titration was performed using an anti-RSV F protein antibody (Millipore, Billerica, MA, USA) and the viral titre was expressed as plaque forming units (PFU). The virus aliquots were stored at −80°C.
RSV clinical isolates preparation
Nasopharyngeal aspirates from two patients were diluted in DMEM low glucose (Gibco by Life Technologies) (5 mL) containing 1000 µg·mL−1 of gentamycin and inoculated into VERO cell culture. After 1 h incubation at 37°C for complete viral adsorption, the viral inoculum was removed and 10 mL of Opti-MEM supplemented with 2% FBS and 1000 µg·mL−1 of gentamycin were added into the flask. The culture was incubated at 37°C with 5% CO2 until the cytopathic effect was visualised. Cells were harvested, centrifuged and the cell pellet was subjected to snap freeze–thaw cycles to obtain new viral particles. Sucrose cryoprotector was added to the viral aliquots and stored at −80°C until titration or further analysis. Lysis plate titration was performed using an anti-RSV antibody and the viral titre was expressed as PFU. The viral nomenclature was made based on the city of isolation/strain number/year (RSV POA10/2018 and RSV POA43/2018). Clinical isolates were assayed by real-time PCR and were identified as follows: RSV POA10/2018 is RSV subtype B and RSV POA43/2018 is RSV subtype A.
Mouse macrophage isolation and stimulation
Alveolar macrophages
AMs were collected by bronchoalveolar lavage (BAL) by flushing the lungs three times with 1 mL of chilled PBS supplemented with EDTA (5 mM). The lavage was repeated twice and AMs from several mice were pooled. Cells were seeded at 3×105·well−1 in AIM-V medium (Gibco by Life Technologies) overnight. Non-adherent cells were then removed by washing, and adherent cells were stimulated with different multiplicity of infections (MOIs) of RSV (0.1–10) for 2, 4 or 6 h at 37°C under 5% CO2. Then, macrophages were labelled with Fixable Viability Dye eFluor 780 (eBioscience by Thermo Fisher Scientific, Waltham, MA, USA), harvested by gentle scraping and the percentage of dead cells was analysed by flow cytometry, as described below.
Peritoneal macrophages
Peritoneal macrophages were obtained 3 days after intraperitoneal instillation of 3 mL of 3% thioglycollate by peritoneal washing with chilled PBS. Cells were seeded at 3×105·well–1 in AIM-V medium overnight. Non-adherent cells were then removed by washing, and adherent cells were stimulated with different MOIs of RSV (0.1–10) for 2, 4 or 6 h at 37°C under 5% CO2. Then, macrophages were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells was analysed by flow cytometry.
Human monocyte isolation and stimulation
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy volunteer donors (mean age of 29 years, from both sexes) who had signed an informed consent form. A total of 20 mL of whole blood was collected and subjected to a Histopaque-1077 (Sigma-Aldrich, Saint Louis, MO, USA) gradient centrifugation for 30 min. PBMCs were seeded at 3×105·well−1 in RPMI 1640 medium (Gibco by Life Technologies) containing 10% FBS overnight. Non-adherent cells were then removed by washing, and adherent cells were infected with RSV at an MOI of 1 for 6 h at 37°C. Then, cells were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells was analysed by flow cytometry.
Cell lines
Human monocyte-like cell line (THP-1; ATCC TIB-202) and promonocytic cell line (U937; ATCC CRL-1593.2) were cultured in RPMI 1640 medium supplemented with 10% FBS. For differentiation to macrophage-like cells, U937 cells were seeded at a density of 2×105·cm−2 in RPMI 1640 with 10% FBS containing phorbol 12-myristate 13-acetate (PMA) (50 ng·mL−1) for 24 h [21]. Human adenocarcinoma alveolar epithelial cell line (A549; ATCC CCL-185) was cultured in DMEM low glucose supplemented with 10% FBS. All cell lines were tested for mycoplasma contamination. THP-1 cells and U937 cells differentiated to macrophage-like cells were infected with RSV at an MOI of 1 for 6 h at 37°C. Cells were then labelled with Fixable Viability Dye eFluor 780 and the percentage of dead cells was analysed by flow cytometry.
TNF measurements
The concentration of TNF in mouse macrophage supernatants and in the supernatants of human monocytes and nasal washes obtained from RSV+ and RSV− infants was determined by ELISA (Sigma-Aldrich), following the manufacturer's instructions.
Lactate dehydrogenase release measurements
Lactate dehydrogenase (LDH) release was detected in macrophage or monocyte supernatants using a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. Readings were carried out at a 490 nm wavelength, using an EZ Read 400 microplate reader (Biochrom, Holliston, MA, USA) and expressed as % LDH release.
Ripk3, Mlkl and Cxcl1 detection by real-time PCR
Mouse lungs or AMs (3×105·well−1) were suspended in Brazol reagent (LGC Biotecnologia, Cotia, Brazil) (1 mL) and the protocol for total RNA extraction was carried out according to the manufacturer's instructions. RNA was reverse-transcribed into cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems by Thermo Fisher Scientific, Waltham, MA, USA) and fluorometric quantification was performed with Qubit (ThermoFisher Scientific). The expression of Ripk3, Mlkl and Cxcl1 was measured using specific primers and probes in a TaqMan Assay (Mm00444947_m1, Mm01244222_m1 and Mm04207460_m1, respectively) using the β-actin gene (Mm02619580_g1) as an endogenous control. Quantification of gene expression was carried out using StepOne (Applied Biosystems by ThermoFisher Scientific). A total of 15 ng of cDNA was used for each reaction. The target gene expression was determined using the 2−ΔCt method (fold change over control expression). The Δ value was calculated by subtracting the cycle threshold (Ct) value for β-actin from the Ct value for Ripk3, Mlkl and Cxcl1 for each sample.
MLKL detection by Western blotting
AMs (3×105·well−1) were infected with RSV (MOI 1) for the indicated times. Cells were then harvested and lysed in RIPA buffer containing a protease inhibitor cocktail. Cell lysates were boiled and subjected to electrophoresis in SDS-polyacrylamide gel (10%) in reducing conditions. The quantified proteins were transferred to a methanol-activated PVDF membrane at 4°C for 2 h. Then, the membranes were blocked with non-fat milk-TBS-T solution for 1 h at room temperature and incubated overnight with anti-MLKL antibodies (1:1000; cat# AP14272, Abgent, San Diego, CA, USA) followed by horseradish peroxidase (HRP) anti-rabbit secondary antibody (1:5000; cat#31460, ThermoFisher Scientific). As an endogenous control, anti-GAPDH antibody (1:5000; cat# MA5-15738, ThermoFisher Scientific), followed by HRP anti-mouse secondary antibody (1:5000; cat# 31430, ThermoFisher Scientific) was used. MLKL expression was detected using the ECL system (Amersham ECL Prime Western Blotting Detection reagent; GE Healthcare Life Sciences, Chicago, IL USA) and the membrane was imaged using the ChemiDoc Imaging System (BioRad, Hercules, CA, USA). Densitometry analysis was performed using ImageJ 1.43 software (US National Institutes of Health, Bethesda, MD, USA). MLKL bands were normalised to GAPDH bands.
Animals and RSV infection
For ex vivo experiments, 8–10-week-old female Balb/c mice were supplied by the breeding facilities of Centro de Modelos Biológicos Experimentais (CeMBE/PUCRS, Porto Alegre, Brazil). Animal procedures were reviewed and approved by the Ethics Committee on Animal Use of PUCRS (CEUA/PUCRS) under protocol number 8049. For in vivo experiments, 8–10-week-old female TNF-α receptor p55 (p55TNF-knock-out or TNF receptor 1 (Tnfr1)−/−) and C57/BL6 wild-type (WT) mice were supplied by the breeding facilities of Centro de Criação de Camundongos Especiais da Faculdade de Medicina de Ribeirão Preto (FMRP/USP, Ribeirão Preto, Brazil), whereas 8–10-week-old female RIPK3−/− and C57/BL6 WT mice were supplied by Vishva Dixit (Genentech, Inc., San Francisco, CA, USA) [22] and housed in the breeding facilities of FMRP/USP. All mice were kept in regular filter-top cages with free access to sterile water and food. Animal procedures were reviewed and approved by the Ethics Committee on Animal Use of the Institute of Biology (protocol numbers 4740-1/2017 and 4740-1(A)/2018) and performed according to the Declaration of Helsinki conventions for the use and care of animals.
For RSV infection, mice were anesthetised with 5% isoflurane and infected intranasally with RSV A2 strain (107 PFU per animal). All animals were weighed daily. Data analysis was performed 5 days post infection. BAL fluid was collected and left lungs were removed after perfusion with formalin for histopathological and immunohistochemistry analysis.
BAL fluid collection
Mice were anesthetised with an intraperitoneal administration of ketamine (0.4 mg·g−1)/xylazine (0.2 mg·g−1) solution and the tracheas were cannulated. The lungs were washed twice with 1 mL of chilled RPMI 1640 medium. BAL fluid was centrifuged, the supernatants collected for cytokine analysis and the pellets suspended for total and differential cell count and flow cytometry. For differential cell counting, the cytospin slides were stained with hematoxylin and eosin (H&E) and the counting procedure was performed by an experienced investigator in a blinded manner.
Histopathological and immunohistochemistry analysis
For the histopathological analysis, the left lungs were embedded in paraffin blocks, cut into 4-µm sections and stained with H&E. The peribronchial and perivascular inflammation was scored according to Barends et al. [23] as absent (0), minimal (1), slight (2), moderate (3), marked (4) or severe (5). Slide analysis was performed in a blinded manner. For immunohistochemistry, the lung sections were submitted to antigen recovery and subsequent labelling with anti-RSV F protein antibody.
AM depletion
AMs were depleted by an intranasal administration of clodronate liposomes (Sigma-Aldrich) (500 µg per animal) or PBS liposomes as a control. Mice were infected with RSV (107 PFU per animal) 24 h later. The animals were weighed daily and data analysis was performed 5 days post infection, as described above. Macrophage depletion was confirmed by flow cytometry analysis in BAL samples.
Flow cytometry
To analyse the percentage cell death following RSV infection in vitro, cells were labelled with Fixable Viability Dye eFluor 780 (1:1000) for 30 min on ice protected from light and then harvested with gentle scraping. Cells were then washed with PBS and cytometry buffer was added. Samples were acquired using a BD FACS Verse (BD Biosciences, San Jose, CA, USA) flow cytometer and analysed using FlowJo software (version 10, Tree Star Inc., San Jose, CA, USA).
Cells isolated from BAL fluid of infected mice were incubated with Mouse Fc Block for 20 min and then labelled with Fixable Viability Dye eFluor 780 (1:1000) for 30 min on ice. Cells were then stained with the surface antibodies anti-CD11c (1:200; clone N418, BioLegend, San Diego, CA, USA), anti-F4/80 (1:200; clone BM8, BioLegend) and anti-CD170 (Siglec-F) (1:200; clone 1RNM44N, eBioscience by ThermoFisher Scientific) for 30 min on ice. Samples were acquired using a BD FACS Verse flow cytometer and analysed using FlowJo software.
Viral load quantitation by real-time PCR
Total lung RNA was extracted and cDNA was synthesised using a GoScript reverse transcriptase kit (Promega), following manufacturer's instructions. The amplification of RSV F protein gene was performed by real-time PCR using the indicated specific primers and probes: forward primer: 5′-AACAGATGTAAGCAGCTCCGTTATC-3′; reverse primer: 5′-GATTTTTATTGGATGCTGTACATTT-3′; and probe: 5′ FAM/TGCCATAGCATGACACAATGGCTCCT-TAMRA/-3′. Primer sequences were synthesised and cloned into pUC57 plasmids (GenScript, Piscataway, NJ, USA) to perform a 10-fold dilution and generate a standard curve for calculation of the viral load. The values obtained from viral copies (based on concentration of the plasmid control) were calculated relative to the weight of the pre-weighed lung portion (copies·g lung−1).
Human samples
Nasopharyngeal lavage samples were obtained from infants hospitalised with bronchiolitis in the first year of life from 2016 to 2018. The samples were from patients who had up to 72 h of clinical signs of a lower respiratory tract infection, such as cough, wheezing and/or respiratory distress. All patients underwent data collection and nasopharyngeal wash to identify respiratory viruses on the first day of hospitalisation. The presence of RSV in patients' samples was identified by both immunofluorescence and RT-PCR. After collection, nasopharyngeal wash samples were diluted in 1 mL saline, homogenised and centrifuged. The supernatant was collected and frozen at −80°C until further analysis.
Ethics statement
For infant sample collection, the parents or legal guardians of all participants signed an informed consent form before sample collection. The study was reviewed and approved by the Research Ethics Committee of PUCRS (CEP/PUCRS) under protocol numbers 1.158.826 and 2.471.678. All procedures followed the standards established by the Declaration of Helsinki.
Statistical analysis
Data are presented as mean±sem. All in vitro experiments were performed in triplicate and repeated at least twice. In vivo experiments were performed twice (n≥3 mice per group in each experiment). The results obtained were analysed using GraphPad Prism 6 statistical software package (GraphPad Software, La Jolla, CA, USA). Comparisons between multiple groups were analysed with one-way ANOVA and a post hoc Tukey or Bonferroni test. When appropriate, unpaired t-test or Mann–Whitney test were employed. The level of significance was set at p≤0.05.
Results
RSV induces necrosis in primary mouse macrophages, human monocytes and macrophage-like cell lines
To evaluate the effect of RSV on AM necrosis, cells were infected with RSV (MOI 1) for 6 h and cell death was assessed by a viability dye that permeates the damaged membrane of dead cells. Our results show that RSV triggered AM necrosis (figure 1a), and that the effect of RSV on macrophage death is dependent on virus replication, given that ultraviolet (UV) inactivation impaired RSV-induced death (figure 1a). To confirm that AMs can be infected by RSV, we quantified the viral load after 6 h of infection. Indeed, RSV was able to infect mouse AMs (figure 1b). Interestingly, RSV was also able to kill peritoneal macrophages (figure 1c) and, again, UV-inactivated RSV failed to trigger cell death (figure 1c), indicating that an active RSV infection is necessary to promote macrophage death. Further characterisation of RSV-induced necrosis revealed that both AM and peritoneal macrophage necrosis occurred in a concentration-dependent manner (supplementary figure S1a, d). Similar results were observed when LDH release was quantified after infection (supplementary figure S1b, e). Cell death could be detected as early as 2 h post infection in AMs (supplementary figure S1c) and 4 h post infection in peritoneal macrophages (supplementary figure S1f). Moreover, we found that RSV was able to induce necrosis in human monocytes (supplementary figure S2a, d), in the human macrophage-like THP-1 cells (supplementary figure S2b, e) and in the human U937 monocytic cells differentiated to macrophage-like cells with PMA (supplementary figure S2c, f). We confirmed that RSV infects and replicates in all these cell types (supplementary figure S2g), and used the human lung epithelial cell line A549 as a positive control for RSV replication (supplementary figure S2g). Finally, we tested the effect of RSV clinical isolates on human monocyte necrosis. Two different RSV clinical isolates were able to promote human monocyte necrosis (figure 1d). Altogether, these data indicate that alveolar and peritoneal mouse macrophages, human monocytes and human macrophage-like cell lines are susceptible to RSV infection-induced necrosis. Furthermore, our results show that clinical RSV strains isolated from patients also trigger necrosis in human monocytes.

Respiratory syncytial virus (RSV) induces necrosis in primary mouse macrophages, human monocytes and macrophage-like cell lines. a) Balb/c mice-derived alveolar macrophages (AMs) (3×105·well−1) were infected with RSV (multiplicity of infections (MOI) 1) or with ultraviolet-inactivated (UV)-RSV (MOI 1) for 6 h at 37°C under 5% CO2. Cells were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells was analysed by flow cytometry. b) AMs (3×105·well−1) were infected with RSV (MOI 1) or left uninfected for 6 h at 37°C with 5% CO2. RNA was harvested and RSV viral load was quantified by real-time PCR. c) Balb/c mouse-derived peritoneal macrophages (3×105·well−1) were infected with RSV (MOI 1) or with UV-RSV (MOI 1) for 6 h. Cells were harvested and labelled with Fixable Viability Dye eFluor 780, and the percentage of dead cells was analysed by flow cytometry. d) Human monocytes (3×105·well−1) were infected with two distinct community isolates of RSV (MOI 1) for 6 h. Cells were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells was analysed by flow cytometry. Data are representative of three independent experiments performed in triplicate and expressed as mean±sem. Data were analysed with one-way ANOVA (a, c, d) or unpaired t-test (b). ND: not detected. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
RIPK1, RIPK3 and MLKL are required for RSV-induced macrophage necroptosis
Most studies on necroptosis commonly stimulate immune and non-immune cells with inflammatory agents in the absence of caspase signalling (using benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-FMK)) [24]. Our results demonstrate that treating macrophages with zVAD-FMK prior to RSV infection did not reduce cell death compared to RSV alone (figure 2a), suggesting that RSV infection does not induce apoptosis and does not require caspase inhibition by zVAD-FMK to induce necrosis in macrophages. To determine the role of RIPK1 on RSV-driven macrophage death, cells were pre-treated with necrostatin-1 (NEC-1), which targets the kinase activity of RIPK1 [25]. Pre-treating macrophages with NEC-1 significantly impaired cell death induced by RSV (figure 2b). The role of RIPK3 on RSV-triggered macrophage death was examined by infecting macrophages from Ripk3−/− mice and littermate control WT mice with RSV for 6 h. Remarkably, Ripk3−/− macrophages were completely resistant to RSV-induced cell death at this time point (figure 2c). Likewise, pharmacological suppression of RIPK3 by GW42X (Synkinase, Parkville, VIC, Australia) [26] protected human monocytes from death promoted by RSV infection (figure 2d), and necrosulfonamide (NSA) (Millipore, Billerica, MA, USA) inhibition of MLKL [14] blocked RSV-triggered human monocyte necroptosis (figure 2e). We next sought to investigate whether the inhibition of necroptosis pathways would impact RSV replication. Indeed, the pharmacological blockade of RIPK1, RIPK3 and MLKL significantly reduced RSV viral loads in human monocytes (figure 2f). Similarly, Ripk3−/− macrophages exhibited a significantly lower viral load when compared to WT macrophages infected with RSV (figure 2g). Interestingly, caspase inhibition by zVAD-FMK did not affect viral load in human monocytes (figure 2f), suggesting that caspases are not involved in virus replication during RSV infection. We then decided to verify whether RSV would induce RIPK3 and MLKL expression in macrophages. RSV infection of macrophages resulted in significant Ripk3 and Mlkl mRNA expression 6 h after infection (figure 2h, i) and an increase in MLKL protein levels at 2 h and 4 h post infection (supplementary figure S3). Taken together, these results indicate that RSV sufficiently promotes mouse macrophage and human monocyte necroptosis through the activation of RIPK1, RIPK3 and MLKL and that necroptosis pathways are detrimental to viral clearance.
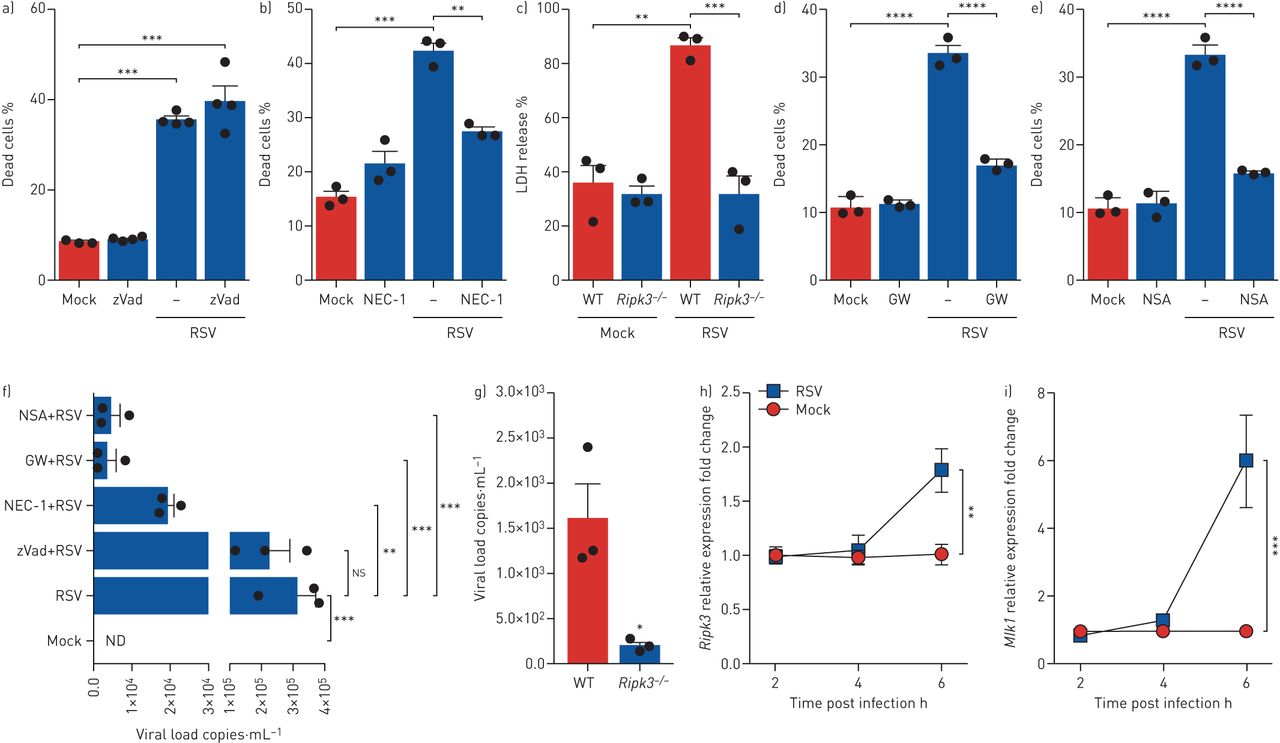
RIPK1, RIPK3 and MLKL are required for respiratory syncytial virus (RSV)-induced macrophage necroptosis. a, b) Balb/c mice-derived macrophages (3×105·well−1) were treated with zVAD-FMK (20 µM) (zVad) (a) or with necrostatin-1 (NEC-1) (30 µM) (b) for 1 h prior to RSV infection (multiplicity of infections (MOI) 1) for 6 h. Macrophages were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells was analysed by flow cytometry. c) Alveolar macrophages (AMs) (3×105·well−1) from Ripk3−/− and littermate control wild-type (WT) mice were infected ex vivo with RSV (MOI 1) for 6 h. Cell death was assessed by lactate dehydrogenase (LDH) release in macrophage supernatants. d, e) Human monocytes (3×105·well−1) were treated with GW42X (GW) (2 µM) (d) or with necrosulfonamide (NSA) (2 µM) (e) for 1 h prior to RSV infection (MOI 1) for 6 h. Cells were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells was analysed by flow cytometry. f) Macrophages were treated with zVAD-FMK (20 µM), NEC-1 (30 µM), GW42X (2 µM) or NSA (2 µM) for 1 h prior to RSV infection (MOI 1) for 6 h. RNA was harvested and the RSV viral load was quantified by real-time PCR. g) Macrophages (3×105·well−1) from Ripk3−/− and littermate control WT mice were infected ex vivo with RSV (MOI 1) for 6 h. RNA was harvested and RSV viral load was quantified by real-time PCR. h, i) AMs were infected with RSV (MOI 1) for 2, 4 or 6 h. RNA was harvested and Ripk3 mRNA (h) and Mlkl mRNA (i) expression was quantified by real-time PCR using the 2−ΔCt method. Data are representative of at least two independent experiments performed in triplicate and expressed as mean±sem. Data were analysed with one-way ANOVA with Tukey's post hoc test (a–f, h, i) or unpaired t-test (g). ns: not significant; ND: not detected. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
RIPK3 exacerbates lung pathology during RSV infection in a manner partially dependent on cell death
It has been previously demonstrated that genetic deletion of RIPK3 protects cIAP2-deficient mice from morbidity and mortality induced by influenza A H1N1 virus infection [17]. Furthermore, Ripk3−/− mice have shown higher survival rates and less weight loss and inflammatory cell infiltration than WT mice when infected with influenza H7N9 virus [18]. Based on the literature and on our findings showing that RSV induces macrophage necroptosis through RIPK3 activation, we sought to investigate the role of RIPK3 during RSV infection in vivo. Ripk3−/− mice were protected from RSV-induced weight loss compared to WT mice (figure 3a) and this was associated with a marked reduction in viral loads in the lung (figure 3b). Ripk3−/− mice also presented with a significant decrease in lung peribronchial inflammatory score compared to WT mice (figure 3c, d). Additionally, RIPK3-deficient mice displayed a significant reduction in the number of macrophages and total cells in BAL fluid (figure 3e). We then asked whether RIPK3 would modulate AM necroptosis during RSV infection by examining the necrotic AM population in BAL samples of Ripk3−/− and WT mice. Ripk3−/− mice exhibited a significant decrease in the frequency of necrotic AMs (CD11c+F4/80+Siglec-F+fixable viability dye+) in BAL fluid following RSV infection when compared to WT mice (figure 3f, g and supplementary figure S4). These results indicate that RIPK3 exacerbates lung pathology during RSV infection in vivo and this is partially coupled to AM death outcomes.

RIPK3 exacerbates lung pathology during respiratory syncytial virus (RSV) infection partially dependent on cell death. Ripk3−/− and littermate control wild-type (WT) mice were infected with RSV (107 PFU·animal−1). Data analysis was performed 5 days post infection. a) Percentage body weight loss post infection relative to initial weight (day 0) (n=5). b) RSV viral load detected in the lung tissue by real-time PCR (n=3). c) Representative images of lung tissue sections stained with hematoxylin and eosin. Scale bars: 200 µm. d) Respective inflammation scores (n=3). e) Total cell number and differential cell counting in bronchoalveolar lavage (BAL) fluid (n=3). f) Percentage of necrotic alveolar macrophages (CD11c+F4/80+Siglec-F+fixable viability dye+) in BAL fluid and g) its representative fluorescence-activated cell sorting profile (n=4–5). Data are representative of two independent experiments and are expressed as mean±sem. Data were analysed with one-way ANOVA with Bonferroni's post hoc test (a) or with Mann–Whitney test (b–e). Neut: neutrophils; Mac: macrophages; Lymph: lymphocytes; Eos: eosinophils; FSCA: forward scatter area. *: p<0.05; **: p<0.01; ***: p<0.001.
AMs play a detrimental role during RSV infection in vivo
The role played by AMs during respiratory viral infections has been a matter of debate. AMs seem to be essential for the early antiviral response against RSV infection [27, 28]. However, depletion of AMs is associated with a significant amelioration of disease, including attenuated weight loss, without altering viral load during RSV reinfection of neonatally infected mice [29]. To establish the role of AMs during RSV infection, we depleted these cells by an intranasal administration of clodronate liposomes 24 h prior to infection in mice. AM depletion was first confirmed by flow cytometry analysis of BAL cells, using anti-CD11c, anti-F4/80 and anti-Siglec-F antibodies recognising mouse AM. A three-fold significant reduction in AM numbers was observed in BAL fluid of mice treated with clodronate liposomes for 24 h compared to PBS liposome-treated mice (supplementary figure S5a, b). After RSV infection, AM-depleted mice demonstrated significantly less weight loss (figure 4a) and a sharp reduction in lung viral loads when compared to non-depleted mice (figure 4b). Corresponding to the decrease in viral load, RSV F protein was not detected by immunohistochemical analysis of lung tissue of RSV-infected AM-depleted mice (figure 4c). Furthermore, AM depletion induced a significant decrease in lung histological inflammatory score in infected mice (figure 4b, e). As expected, AM-depleted mice displayed a lower number of macrophages in BAL fluid but, surprisingly, a greater number of neutrophils (figure 4f). As a marker of cell damage, we measured LDH activity in BAL samples of mice. We were able to detect increased levels of LDH in BAL fluid of RSV-infected mice compared to uninfected ones; however, the treatment with clodronate liposomes did not affect LDH release in mice (figure 4g). Interestingly, AM depletion abolished RSV-induced upregulation of Ripk3 and Mlkl mRNA in the lung tissue (figure 4h, i). Altogether, these data suggest that AMs play a harmful role during RSV infection and that AMs account for most RIPK3 and MLKL expression in the lung tissue after infection.
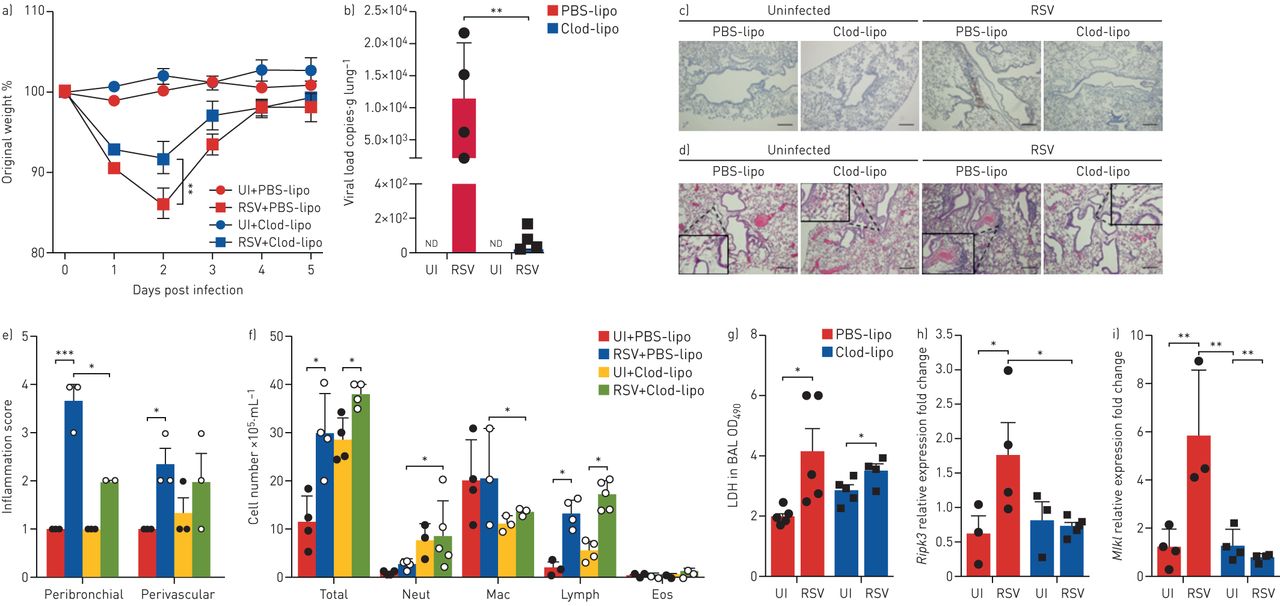
Alveolar macrophages play a detrimental role during respiratory syncytial virus (RSV) infection in vivo. Balb/c mice were depleted of alveolar macrophages by intranasal administration of clodronate liposomes (Clod-lipo) (500 µg·animal−1) or PBS liposomes (PBS-lipo). Mice were infected with RSV (107 PFU·animal−1) 24 h later. Data analysis was performed 5 days post infection. a) Percentage of body weight loss post infection relative to initial weight (day 0) (n=5). b) RSV viral load detected in the lung tissue by real-time PCR (n=4–5). c) Representative images of immunohistochemical staining of lung tissues using an anti-RSV fusion protein antibody. Scale bars: 200 µm. d) Representative images of lung tissue sections stained with hematoxylin and eosin. Scale bars: 200 µm. e) Peribronchial and perivascular inflammation scores respective to histological images shown in d (n=3). f) Total cell number and differential cell counting in bronchoalveolar lavage (BAL) fluid (n=4–5). g) Lactate dehydrogenase (LDH) activity measured at an optical density of 490 nm (OD490) in BAL samples (n=4–5). h, i) Ripk3 mRNA (h) and Mlkl mRNA (i) expression in the lung tissue quantified by real-time PCR using the 2−ΔCt method and expressed as fold change (n=3–4). Data are representative of two independent experiments and are expressed as mean±sem. Data were analysed with one-way ANOVA with Tukey's post hoc test. UI: uninfected; ND: not detected; Neut: neutrophils; Mac: macrophages; Lymph: lymphocytes; Eos: eosinophils. *: p<0.05; **: p<0.01; ***: p<0.001.
Autocrine TNF mediates RSV-induced macrophage necroptosis in vitro
We have recently shown that macrophages respond to ex vivo RSV infection by releasing TNF [30]. Because TNF has been extensively studied as a necroptotic trigger [31], we sought to elucidate the role of TNF in RSV-induced macrophage necroptosis. RSV elicited the secretion of TNF by macrophages in a concentration-dependent manner (figure 5a), but only the replicating virus was able to induce TNF release (figure 5b). To explore whether TNF plays a role in macrophage necroptosis triggered by RSV, we treated the cells with a therapeutic TNF-specific neutralising antibody (infliximab), a chimeric (mouse/human) antibody that cross-reacts with murine TNF [32]. Macrophages were treated with infliximab immediately before RSV infection. Neutralisation of TNF with infliximab abolished RSV-induced macrophage necroptosis in vitro (figure 5c). Interestingly, blocking TNF secretion with brefeldin A, a well-known protein transport inhibitor [33], decreased TNF concentrations in macrophage supernatants to the control levels (mock) (supplementary figure S6a) and profoundly reduced macrophage death caused by RSV (supplementary figure S6b). We then infected macrophages from Tnfr1−/− and littermate control WT mice with RSV ex vivo. Notably, Tnfr1−/− macrophages were completely protected from RSV-induced death (figure 5d). Next, we sought to further elucidate the role of necroptosis pathways on TNF release induced by RSV infection. The pharmacological inhibition of RIPK1, RIPK3 and MLKL significantly reduced TNF secretion promoted by RSV in human monocytes (figure 5e), suggesting that necroptosis pathways are necessary for RSV-elicited TNF release. Last, we observed that the infection of human monocytes with two different RSV clinical isolates was able to induce TNF release (figure 5f). Collectively, these results indicate that RSV-triggered macrophage necroptosis is mediated by autocrine TNF and that necroptosis pathways are involved in TNF release.

Autocrine tumor necrosis factor (TNF) mediates respiratory syncytial virus (RSV)-induced macrophage necroptosis in vitro. a, b) Balb/c mice-derived macrophages (3×105·well−1) were infected with increasing multiplicity of infections (MOIs) of RSV (0.1–5) for 6 h (a) or with active or ultraviolet-inactivated RSV (UV-RSV) (MOI 1) for 6 h (b). Supernatants were collected and TNF concentrations measured by ELISA. c) Balb/c mice-derived macrophages (3×105·well−1) were infected with RSV (MOI 1) in the presence of infliximab (IFX) (10 µg·mL−1) for 6 h. Cells were labelled with Fixable Viability Dye eFluor 780, harvested and the percentage of dead cells analysed by flow cytometry. d) Alveolar macrophages (3×105·well−1) from Tnfr1−/− and littermate control wild-type (WT) mice were infected ex vivo with RSV (MOI 1) for 6 h. Cell death was assessed by lactate dehydrogenase (LDH) release in macrophage supernatants. e) Human monocytes (3×105·well−1) were treated with necrostatin-1 (NEC-1) (30 µM), GW42X (GW) (2 µM) or necrosulfonamide (NSA) (2 µM) for 1 h prior to RSV infection (MOI 1) for 6 h. Cell supernatants were collected and TNF concentrations measured by ELISA. f) Human monocytes (3×105·well−1) were infected with two distinct community isolates of RSV (MOI 1) for 6 h. Cell supernatants were collected and TNF concentrations measured by ELISA. Data are representative of at least two independent experiments performed in triplicate and expressed as mean±sem. Data were analysed with one-way ANOVA with Tukey's post hoc test. *: p<0.05; ***: p<0.001; ****: p<0.0001.
Lack of TNFR1 signalling ameliorates RSV infection-induced pathology and diminishes AM necroptosis
Based on our findings that autocrine TNF mediates RSV-induced macrophage necroptosis ex vivo, we were interested in elucidating the role of TNF in RSV infection and in RSV-induced AM necroptosis in vivo. Therefore, Tnfr1−/− and littermate WT mice were infected with RSV. Our results demonstrate that Tnfr1−/− mice were protected from RSV-induced body weight loss (figure 6a) and displayed significantly lower viral loads in the lung tissue (figure 6b) when compared to WT mice. Moreover, Tnfr1−/− mice presented with decreased histological peribronchial and perivascular inflammation scores in comparison to WT mice (figure 6c). The number of total leukocytes, as well as the number of macrophages, in BAL fluid of Tnfr1−/− mice was significantly lower after RSV infection than in WT mice (figure 6d). Interestingly, the expression of Cxcl1 mRNA in the lung tissue of Tnfr1−/− mice was profoundly impaired (figure 6e), confirming that inflammation was dampened during RSV infection in these mice. Similarly, the expression of Ripk3 and Mlkl transcripts was significantly reduced in Tnfr1−/− mice (figure 6f, g), suggesting that TNF mediates the expression of RIPK3 and MLKL in the lung tissue following RSV infection. Finally, to determine whether the lack of TNFR1 signalling would influence AM necroptosis during RSV infection, we analysed the necrotic AM population in BAL samples of Tnfr1−/− and WT mice. Remarkably, Tnfr1−/− mice displayed a six-fold decrease in the frequency of necrotic AMs (CD11c+F4/80+Siglec-F+fixable viability dye+) in BAL fluid following RSV infection when compared to WT mice (figure 6h and supplementary figure S7). Altogether, these data strongly suggest that TNFR1 signalling mediates lung pathology and AM necroptosis triggered by RSV infection.

Lack of tumor necrosis factor receptor 1 (TNFR1) signalling ameliorates respiratory syncytial virus (RSV) infection-induced pathology and diminishes alveolar macrophage necroptosis. Tnfr1−/− and littermate control wild-type (WT) mice were infected with RSV (107 PFU·animal−1). Data analysis was performed 5 days post infection. a) Percentage of body weight loss post infection relative to initial weight (day 0) (n=5). b) RSV viral load detected in the lung tissue by real-time PCR (n=4). c) Representative images of lung tissue sections stained with hematoxylin and eosin and its respective inflammation scores (n=3). Scale bars: 200 µm. d) Total cell number and differential cell counting in bronchoalveolar lavage (BAL) fluid (n=3). e–g) Cxcl1 mRNA (e), Ripk3 mRNA (f) and Mlkl mRNA (g) expression in the lung tissue quantified by real-time PCR using 2−ΔCt method (n=3). h) Percentage of necrotic alveolar macrophages (CD11c+F4/80+Siglec-F+fixable viability dye+) in BAL fluid and its representative fluorescence-activated cell sorting profile (n=4). Data are representative of two independent experiments and are expressed as mean±sem. Data were analysed with unpaired t-test. Neut: neutrophils; Mac: macrophages; Lymph: lymphocytes; Eos: eosinophils; FSCA: forward scatter area. *: p<0.05; **: p<0.01; ***: p<0.001.
Elevated TNF levels in nasal wash correlate with disease severity in infants with RSV bronchiolitis
We assessed the contribution of TNF during acute RSV infection by measuring TNF levels in nasal wash samples from infants with RSV bronchiolitis. There were no significant differences in age (p=0.6784), sex (p=0.1507) or birth weight (p=0.4484) among patients from the RSV+ group compared to the RSV− group (supplementary table S1). The concentrations of TNF were significantly greater (p=0.0014) in the nasal samples from RSV+ infants (209.41±23.55 pg·mL−1, n=39) compared to RSV− infants (113.38±14.16 pg·mL−1, n=5) (figure 7a). We next analysed the levels of TNF with respect to the need for supplemental oxygen therapy. There was a trend towards a positive correlation between the levels of TNF and the need for oxygen therapy measured in days, although it was nonsignificant (p=0.0649) (figure 7b). In contrast, there was a significant positive correlation between TNF levels in nasal wash and the length of hospital stay (p=0.0010) (figure 7c). Interestingly, the infants who had a longer hospital stay (≥7 days) exhibited greater concentrations of TNF in the nasal wash (341.6±60.24 pg·mL−1) than those with a shorter (<7 days) hospital stay (119.7±17.93 pg·mL−1) (p=0.0002) (figure 7d). These data suggest that elevated TNF levels in the nasal wash positively correlate with disease severity in infants with RSV infection.

Elevated tumor necrosis factor (TNF) levels in nasal wash correlate with disease severity in infants with respiratory syncytial virus (RSV) bronchiolitis. Nasal wash samples were collected from infants up to 12 months of age who were admitted to the hospital with acute bronchiolitis. The presence of RSV was confirmed by real-time PCR. a) TNF levels were measured by ELISA in the supernatants of nasal wash and compared among infants positive for RSV (RSV+) and infants negative for RSV (RSV−). b) Spearman linear correlation between TNF levels and time of O2 need of RSV+ infants with acute bronchiolitis. c) Spearman linear correlation between TNF levels and length of hospital stay of RSV+ infants with acute bronchiolitis. d) Comparison of TNF levels in RSV+ infants who spent <7 days in hospital with those who spent ≥7 days in hospital. Data are expressed as mean±sem and were analysed with an unpaired t-test (a, d). **: p=0.0014; ***: p=0.0002.
Discussion
Respiratory viruses have been shown to trigger necroptosis in different cell types [16, 34–36]. Accordingly, recent studies have reported that RSV induces necroptosis in alveolar epithelial cells and THP-1 cells [19, 20]. However, whether RSV is able to trigger necroptosis in primary macrophages, important cells for the immune response against this viral infection, has not been investigated. In this study, we demonstrate that RSV infection induces alveolar and peritoneal mouse macrophage necroptosis in a concentration- and time-dependent manner. Moreover, RSV replicative activity was essential for necroptosis induction, given that UV-inactivated RSV failed to trigger macrophage death. We extended our findings by showing that human monocytes, THP-1 cells and U937 cells differentiated to macrophage-like cells are all susceptible to RSV infection-triggered necroptosis. Importantly, in an effort to use a clinically representative strain of RSV, we infected human monocytes with two distinct RSV community isolates and we demonstrate that these isolates were able to promote necroptosis.
Necroptosis can occur when caspase-8 activity is compromised, and it is well known that virus-encoded caspase-8 inhibitors may switch the cell death programme to necroptosis, as is the case for vaccinia virus. Vaccinia virus-infected cells are susceptible to TNF-induced, RIPK1- and RIPK3-dependent necroptosis owing to the expression of B13R, a caspase-8 inhibitor [15, 37, 38]. To date, there is no description in the literature of an RSV-encoded caspase-8 inhibitor. Thus, in an attempt to inhibit caspase-8 activity, we treated macrophages with the pan-caspase inhibitor zVAD-FMK prior to RSV infection. However, in our setting, zVAD-FMK was unable to further increase macrophage necroptosis induced by RSV, indicating that RSV infection is sufficient to induce necroptosis. Our data are supported by previous findings showing that RSV naturally triggers alveolar epithelial cell necroptosis [20].
The interaction of RIPK1 and RIPK3 through the RIP homotypic interaction motif (RHIM)–RHIM domains is central to necroptotic signalling and leads to necrosome formation [39], which results in the mutual phosphorylation and activation of RIPK1 and RIPK3 [40]. RIPK3 activation causes the phosphorylation of its substrate, MLKL, which in turn oligomerises and translocates to the plasma membrane, where it forms membrane-disrupting pores, leading to cell lysis [41–44]. To characterise the requirement of RIPK1 during RSV-elicited AM necroptosis, we treated the cells with NEC-1, which blocks the kinase activity of RIPK1 [25]. Treating macrophages with NEC-1 significantly decreased cell death induced by RSV. Furthermore, we have shown that RIPK3 is essential for RSV-triggered necroptosis, given that Ripk3−/− macrophages were completely resistant to cell death promoted by the virus and the pharmacological suppression of RIPK3 practically abolished human monocyte necroptosis caused by RSV infection. Likewise, NSA inhibition of MLKL profoundly impaired RSV-incited human monocyte necroptosis. Importantly, we found that RSV induced both Ripk3 and Mlkl mRNA expression at 6 h post infection. We also observed that the pharmacological inhibition or genetic deletion of necroptosis pathways markedly reduced viral loads in AMs and human monocytes, suggesting that necroptosis promotes RSV persistence in these cells. Consistent with our findings, RIPK3 has been shown to facilitate coxsackievirus replication in intestinal epithelial cells [45]. Therefore, AMs and human monocytes clearly rely on RIPK1, RIPK3 and MLKL to undergo cell death and these molecules act as positive regulators of RSV replication.
Based on the importance of RIPK3 for in vitro RSV-induced macrophage necroptosis, we assumed that RIPK3 would be crucial for RSV-induced disease in vivo. In fact, the genetic deficiency of RIPK3 protected mice from RSV-induced weight loss and reduced the viral load in the lungs. Additionally, Ripk3−/− mice displayed impaired recruitment of macrophages to the lungs, which may be secondary to the reduction of viral replication in the absence of RIPK3. Thus, we provide evidence for a detrimental role of RIPK3 during RSV infection. Similarly, RIPK3 deficiency protected mice from infection with influenza H7N9 virus by decreasing weight loss and the infiltration of inflammatory cells in the lungs [18]. It has previously been shown that RIPK3 simultaneously activates both MLKL-driven necroptosis and Fas-associated protein with death domain (FADD)-mediated apoptosis to protect against the spread of influenza A H1N1 virus infection [16]. However, RIPK3 has been reported to promote inflammation and regulation of viral replication without triggering cell death [46]. In our setting, RIPK3 exacerbated lung injury by promoting inflammation and AM necroptosis, to some extent. Hence, the physiological relevance of RIPK3 during a viral infection may be determined by the virus subtype and likely interfering factors in the host immune response. As an important component for necrosome formation, RIPK1 is also detrimental during viral bronchiolitis in mice, given that both pharmacological blockade and genetic deficiency of RIPK1 protected mice from weight loss, neutrophilic inflammation and alarmin release [20]. Therefore, RIPK1 and RIPK3 are pathological players during RSV infection.
The impact that AMs have on respiratory virus-induced disease is controversial. While AMs seem to be essential for the early antiviral response against RSV infection [27, 28, 47, 48] and to prevent lethal influenza pneumonia [49], the depletion of AMs is associated with a significant amelioration of disease, including attenuated weight loss and decreased airway obstruction, following pulmonary coronavirus and human metapneumovirus infection in mice [27, 50]. Moreover, AM depletion prolonged survival in response to acute pneumovirus infection in mice and reduced weight loss without altering viral load during RSV reinfection of neonatally infected mice [29, 51]. Our data clearly demonstrate that AMs enhance RSV replication in the lungs and significantly contribute to clinical disease and lung pathology. These findings were consistently underlined by the reduction in lung viral loads, attenuated weight loss and impaired peribronchial inflammation score in AM-depleted mice. Thus, it is reasonable to assume that AMs may represent the first immune cell target of RSV infection in the lungs, acting as virus factories in the early phase of disease. Importantly, AM depletion abolished the RSV-induced upregulation of Ripk3 and Mlkl mRNA, suggesting that AMs account for most of the RIPK3 and MLKL expression in the lung tissue after infection, prompting necroptosis.
Because RSV-induced macrophage necroptosis required virus replication, we hypothesised that during the RSV replication cycle the necroptotic machinery was activated indirectly, e.g. through the autocrine activity of a virus-induced pro-necroptotic cytokine such as TNF. Consistent with this assumption, we have recently shown that macrophages respond to RSV infection by secreting TNF [30]. We extended these findings and show that RSV triggers the release of TNF in a concentration-dependent manner and that TNF is released as a result of RSV replication, because UV-inactivated RSV was not able to induce TNF secretion from macrophages. We then investigated whether TNF would mediate RSV-promoted macrophage necroptosis. The neutralisation of TNF with infliximab, a therapeutic anti-TNF antibody, abolished RSV-induced macrophage necroptosis. Interestingly, when TNF secretion was blocked by brefeldin A, the RSV effect on macrophage death was lost. Moreover, Tnfr1-deficient macrophages were completely resistant to RSV-triggered cell death. Thus, our findings establish that TNF is secreted during RSV replication in AMs, which, in turn, promotes macrophage necroptosis in an autocrine manner. Furthermore, it has recently been shown that RIPK3 activation leads to cytokine production even after loss of plasma membrane integrity [52]. Hence, we decided to clarify whether RIPK1, RIPK3 and MLKL would promote TNF synthesis in the context of RSV-induced necroptosis. The pharmacological blockade of necroptosis proteins markedly impaired RSV-induced TNF secretion. These data imply that besides inducing TNF production through its replicative activity in macrophages, RSV also leads to TNF synthesis by activating the necroptotic machinery even when macrophages are committed to cell death. The secretion of TNF by necroptosis activation could lead to a positive feedback loop, worsening lung pathology induced by RSV infection. In line with our findings, influenza virus, which also elicits RIPK3-mediated necroptosis [16, 35], has been reported to promote such a phenomenon [52].
Given our observation on the participation of TNF during in vitro RSV infection of AMs, we sought to elucidate the role of TNF on RSV infection and on RSV-induced AM necroptosis in vivo. In our setting, the lack of TNFR1 signalling was protective against RSV-induced weight loss and lung inflammation. In addition, Tnfr1−/− mice presented a sharp reduction in viral loads and in the infiltration of macrophages in the lungs. In accordance, mice lacking both TNF and interleukin 1 (IL-1) receptors exhibit decreased lung inflammation and a significant delay in mortality caused by a lethal challenge with H5N1 virus [53]. Furthermore, TNF and macrophages are critical to RSV-induced asthma exacerbations in mice [54]. We found that the expression of Ripk3 and Mlkl was significantly impaired in the lungs of Tnfr1−/− mice following RSV infection, which may reflect the profound reduction in the percentage of necrotic AMs in the absence of TNFR1 signalling. We provide compelling evidence that TNF is critically involved in the severity of clinical disease during RSV infection by increasing viral load and lung pathology, as well as promoting RIPK3- and MLKL-dependent AM necroptosis. In support of our findings, it has been previously demonstrated that TNF-induced necroptosis requires the activation of RIPK1, RIPK3 and MLKL in different cell types and in mouse models [41, 55, 56]. In addition, RIPK1-RIPK3-mediated cellular damage by necroptosis drives mortality during TNF-induced systemic inflammatory response syndrome [57].
Previous studies have shown that TNF, together with other pro-inflammatory cytokines, is elevated in the nasal wash of infants infected with RSV [58–60]. Corroborating with the previous findings, our results demonstrate that RSV-infected infants exhibit higher levels of TNF in the nasal wash than infants infected with other respiratory viruses. TNF signalling within the respiratory tract governs different pathophysiological functions of the airways, including inflammatory cell recruitment and activation to pathogens [61]. A possible mechanism by which high levels of TNF may increase disease severity is through the upregulation of adhesion molecules on small capillaries, thereby inducing the recruitment of inflammatory cells, e.g. neutrophils, macrophages and eosinophils, into the airways [62]. It has also recently been reported that TNF is involved in bronchoconstriction during RSV infection in mice [63]. We measured disease severity in infants on the basis of the length of hospital stay and the duration of supplemental oxygen therapy. We were not able to find a positive correlation between the levels of TNF and the need for oxygen therapy; however, we did find a significant positive association between TNF levels in nasal wash and the length of hospital stay. We found that infants who stayed ≥7 days in the hospital presented with greater concentrations of TNF in the nasal wash than those who stayed <7 days in the hospital. Collectively, these data suggest that elevated TNF levels in the nasal wash positively correlate with disease severity in infants with RSV infection. Moreover, it has previously been shown that RSV bronchiolitis is associated with TNF haplotypes [64], suggesting that the genetically mediated upregulation of TNF might support exaggerated inflammation in the airways and thus promote a more severe course of RSV infection.
In conclusion, our data demonstrate that autocrine TNF mediates RIPK1-, RIPK3- and MLKL-dependent AM necroptosis triggered by RSV infection and that necroptosis pathways are detrimental to viral clearance. Moreover, TNF-mediated AM necroptosis plays an important role in the pathogenesis of RSV infection (figure 8). We provide evidence that elevated nasal levels of TNF are associated with disease severity in infants with RSV bronchiolitis. We propose that targeting TNF and/or the necroptotic machinery may be valuable as an additional therapeutic approach to reduce the respiratory morbidity caused by RSV infection in young children and babies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanisms of respiratory syncytial virus (RSV)-induced macrophage necroptosis. During RSV infection in macrophages, tumor necrosis factor (TNF) is produced and secreted as a result of viral replication and activation of the necroptotic machinery. In an autocrine manner, TNF binds to TNF receptor 1 (TNFR1), leading to receptor-interacting protein kinase 1 (RIPK1) and RIPK3 phosphorylation. As a consequence of RIPK3 activation, mixed-lineage kinase domain-like (MLKL) is phosphorylated and activated, leading to macrophage necroptosis. In mice, RSV-induced TNF-mediated alveolar macrophage necroptosis results in weight loss, increased lung viral loads and exacerbated lung inflammation.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-03764-2020.SUPPLEMENT
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-03764-2020.Shareable
Acknowledgements
We thank Géssica Antunes, Deise do Nascimento de Freitas, Rodrigo Dornelles, Janaína Pasetti Nunes (all from PUCRS) and Rosemeire Florença de Paula (UNICAMP) for their excellent technical assistance. We also thank the Rheumatology Service of São Lucas Hospital (Porto Alegre, Rio Grande do Sul, Brazil) for kindly donating infliximab (Remicade; Centocor, Horsham, PA, USA).
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: B.N. Porto conceived the study. B.N. Porto and M.A.R. Vinolo designed and supervised the experiments. B.N. Porto and L.D. Santos wrote the manuscript with input from M.A.R. Vinolo, R. Weinlich and A.P. Duarte de Souza. L.D. Santos, K.H. Antunes, S.P. Muraro, G.F. de Souza, A.G. da Silva, J.S. Felipe, L.C. Zanetti, R.S. Czepielewski and K. Magnus performed the experiments and analysed the data. F. Maito performed the lung histopathological and immunohistochemistry analysis. M. Scotta, R. Mattiello and A.P. Duarte de Souza provided the patients’ nasal washes and contributed to the data analysis. All authors reviewed and commented on the manuscript.
Conflict of interest: L.D. Santos reports grants from CAPES, during the conduct of the study.
Conflict of interest: K.H. Antunes has nothing to disclose.
Conflict of interest: S.P. Muraro has nothing to disclose.
Conflict of interest: G.F. de Souza has nothing to disclose.
Conflict of interest: A.G. da Silva has nothing to disclose.
Conflict of interest: J.S. Felipe has nothing to disclose.
Conflict of interest: L.C. Zanetti has nothing to disclose.
Conflict of interest: R.S. Czepielewski has nothing to disclose.
Conflict of interest: K. Magnus has nothing to disclose.
Conflict of interest: M. Scotta has nothing to disclose.
Conflict of interest: R. Mattiello has nothing to disclose.
Conflict of interest: F. Maito has nothing to disclose.
Conflict of interest: A.P. Duarte de Souza has nothing to disclose.
Conflict of interest: R. Weinlich has nothing to disclose.
Conflict of interest: M.A.R. Vinolo has nothing to disclose.
Conflict of interest: B.N. Porto reports grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), during the conduct of the study.
Support statement: This study was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) grant number 456282/2014-9 (to B.N. Porto) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) – Finance Code 001. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received October 7, 2020.
- Accepted November 27, 2020.
- Copyright ©ERS 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










