





Abstract
Oxidised phosphatidylcholines (OxPCs) are produced under conditions of elevated oxidative stress and can contribute to human disease pathobiology. However, their role in allergic asthma is unexplored. The aim of this study was to characterise the OxPC profile in the airways after allergen challenge of people with airway hyperresponsiveness (AHR) or mild asthma. The capacity of OxPCs to contribute to pathobiology associated with asthma was also to be determined.
Using bronchoalveolar lavage fluid from two human cohorts, OxPC species were quantified using ultra-high performance liquid chromatography-tandem mass spectrometry. Murine thin-cut lung slices were used to measure airway narrowing caused by OxPCs. Human airway smooth muscle (HASM) cells were exposed to OxPCs to assess concentration-associated changes in inflammatory phenotype and activation of signalling networks.
OxPC profiles in the airways were different between people with and without AHR and correlated with methacholine responsiveness. Exposing patients with mild asthma to allergens produced unique OxPC signatures that associated with the severity of the late asthma response. OxPCs dose-dependently induced 15% airway narrowing in murine thin-cut lung slices. In HASM cells, OxPCs dose-dependently increased the biosynthesis of cyclooxygenase-2, interleukin (IL)-6, IL-8, granulocyte−macrophage colony-stimulating factor and the production of oxylipins via protein kinase C-dependent pathways.
Data from human cohorts and primary HASM cell culture show that OxPCs are present in the airways, increase after allergen challenge and correlate with metrics of airway dysfunction. Furthermore, OxPCs may contribute to asthma pathobiology by promoting airway narrowing and inducing a pro-inflammatory phenotype and contraction of airway smooth muscle. OxPCs represent a potential novel target for treating oxidative stress-associated pathobiology in asthma.
Abstract
Unique profiles of oxidised phospholipids in the human lung correlate with airway pathophysiology. They are novel pro-inflammatory mediators with direct effects in structural cells via complex pathways, and are not targeted by standard asthma therapies. https://bit.ly/34UO2AL
Introduction
Oxidative stress is a major feature of asthma pathophysiology, but it is not targeted by current treatments. Chronic inflammation involving neutrophils, eosinophils and alveolar macrophages and exposure to environmental pollutants, allergens and oxidants in cigarette smoke contribute to the production of reactive oxygen species (ROS) that can overwhelm the body's antioxidant mechanisms [1]. ROS, such as superoxide and hydrogen peroxide, and other biomarkers of oxidative stress (e.g. 8-isoprostane, exhaled nitric oxide and lipid peroxides) are associated with the severity of asthma and airway hyperresponsiveness (AHR), airway mucus secretion and inflammation, emergency department visits and corticosteroid insensitivity [2–6]. People with severe asthma and chronic obstructive pulmonary disease have decreased plasma levels of antioxidant enzymes such as superoxide dismutase and glutathione peroxidase, and reduced plasma glycine and glutamine, which is needed to synthesise the antioxidant glutathione [7]. Despite the evidence that ROS play a key role in asthma severity, we have no effective therapies that directly target oxidative stress in asthma; thus, ROS-associated pathobiology is a persistent threat to disease management.
ROS oxidise and damage DNA, as well as modifying proteins and lipids, leading to bio-activation of otherwise inert homeostatic molecules [8]. The airway lining is particularly susceptible to the effects of ROS owing to its large surface area and contact with environmental pollutants and atmospheric oxygen. ROS generated in the airways can oxidise the phospholipid-rich lining of the airways, including the pulmonary surfactant [9], resulting in the production of bioactive oxidised phospholipids. Phosphatidylcholine, the most abundant phospholipid in mammals [10], can undergo oxidation of the sn-2 polyunsaturated fatty acid chain, with subsequent fragmentation or cyclisation to generate hundreds of different oxidised phosphatidylcholines (OxPCs) (Box 1) [9]. OxPCs alter biological membrane structure and function, and activate signalling pathways that exacerbate and drive oxidative stress and persistent inflammation, thus promoting ageing and disease pathogenesis, e.g. atherosclerosis and renal ischaemia reperfusion injury [11–14]. OxPCs are also causally linked to acute lung injury and acute respiratory distress syndrome, and regulate antimicrobial mechanisms in sepsis [13, 15]. Despite the potential for OxPCs to contribute to inflammatory disease, there is little information about whether they are present in and contribute to asthma pathobiology. In this study, we used human cohort data and human cell culture models to reveal that OxPCs are present in airways, correlate with measures of AHR and lung dysfunction after allergen challenge, induce a pro-inflammatory phenotype in HASM cells and promote airway constriction in murine lung slices.
BOX 1 Abbreviation list for phosphocholines
| HDiA-PC | 4-hydroxy-7-carboxyhex-5-enoic acid ester of 1-stearoyl-2-linoleoyl-sn-glycero-phosphocholine |
| KODA-PPC | 1-palmitoyl-2-(9,12-dioxododec-10-enoyl)-sn-glycero-3-phosphocholine |
| KDiA-PC | 1-(palmitoyl)-2-(4-keto-dodec-3-ene-dioyl)phosphocholine |
| PAPC | 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine |
| PAPC-keto | 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine ketone |
| PGPC | 1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine |
| PLPC-OH | 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine hydroxide |
| PLPC-OOH,OH | 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine hydroxy aldehyde |
| PSPC | 1-palmitoyl-2-stearoyl-sn-glycero-3-phosphocholine |
| SGPC | 1-stearoyl-2-glutaroyl-sn-glycero-3-phosphocholine |
Materials and methods
Detailed methods can be found in the supplementary material.
Human experiments
By taking advantage of existing samples from two established human cohorts available through the Canadian Respiratory Research Network, we were able to explore changes in the OxPC profile of the lungs associated with AHR or allergen exposure. We set out to identify the OxPC profile in subjects with or without AHR, and in subjects with asthma after allergen challenge. The DE3 cohort (Diesel Exhaust 3, Vancouver, BC) comprises subjects with (n=8) and without (n=5) AHR to methacholine [16]. These are otherwise healthy individuals with no smoking history and without a diagnosis of airways disease; thus, none of these subjects was on inhaled corticosteroid therapy (table 1). Following characterisation of methacholine responsiveness, human bronchoalveolar lavage fluid (BALF) samples were collected from the 13 participants by bronchoscopy [16]. The DC cohort (Hamilton, Ontario) is a second study group comprising non-smoking subjects with mild atopic asthma (n=10) who are not on inhaled corticosteroids or long-acting β2 agonists (table 2) [17]. BALF samples were collected 24 h after allergen or diluent challenge and filtered (40 µm mesh filter). Cell-free supernatants were then stored at −80°C for analysis of oxidised phospholipids [18]. Together, these cohorts had unique features that enabled us to address critical issues, including the capacity to assess OxPCs in BALF from atopic asthmatic subjects in direct response to allergen challenge (DC cohort); test for an association between BALF OxPCs and clinically validated AHR (DC and DE3 cohorts); and discriminate whether BALF OxPCs are associated with airway dysfunction, independent of asthma diagnosis (DE3 cohort).
Patient characteristics for DC cohort (Hamilton, ON, Canada)
Patient characteristics for DE3 cohort (Vancouver, BC, Canada)
Oxidised phospholipid quantification
OxPC content in BALF from humans was extracted using the Folch method, then identified and quantified by ultra-high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) using multiple reaction monitoring standards as previously described [14].
Cell culture
Primary human airway smooth muscle (HASM) cells (passage 4–6) isolated from lung tissue resections were grown to confluence and then serum starved for 7 days in the presence of insulin-transferrin-selenium to induce a phenotype that partially resembled HASM cells in vivo [19–21]. For experiments, cultures were exposed to increasing concentrations of oxidised (Ox) PAPC for 24 h unless otherwise indicated.
Lipid peroxidation
25 mg of PAPC and PSPC were oxidised in room air for 4 days and then re-suspended at a concentration of 1 mg·mL−1 in chloroform:methanol (2:1). A small sample of OxPAPC was tested for degree of oxidation using an HPLC/electrospray ionisation-MS/MS system as previously described [14]. This OxPAPC represented a mixture of OxPC species that could be used to recapitulate the OxPC profile seen in human samples.
Cytokine analysis
Cytokine analysis on cell culture supernatants was performed using multiplex arrays from Eve Technologies (Alberta, Canada) and Mesoscale Discovery Platform (Rockville, MD, USA).
Quantitative PCR
Cyclooxygenase-2 (COX2) mRNA abundance was quantified using quantitative PCR (qPCR) on RNA from HASM cells. Housekeeping genes used were YWHAZ, UBC and GAPDH.
Western blotting
10 µg of total protein isolated from HASM cells was run on a 10% gel, blotted onto nitrocellulose, and probed with COX2 and COX1 antibodies.
Oxylipin quantification
Cell supernatant, collected in phenol-free DMEM, was analysed using HPLC-MS/MS as previously described [22].
Kinome array
Cell lysates collected from cells exposed to OxPC 80 µg·mL−1 for 1, 3 or 6 h were collected in phosphoarray lysis buffer and run on a peptide array as previously described [23] to quantify the OxPC-induced cellular kinome. Specifically, lysates were run on human kinome peptide arrays (JPT Technologies, Berlin, Germany), which comprise 308 unique kinase recognition sites. Results were visualised using PRO-Q Diamond Phosphoprotein stain and subsequently scanned on a PowerScanner microarray scanner (Tecan, Morrisville, NC, USA) with a 580 nm filter to detect fluorescence. Signal intensity values were collected with Array-Pro Analyser version 6.3 software (Media Cybernetics, Rockville, MD, USA).
Thin-cut lung slice studies
Lungs from 8–10 week-old female BALB/c mice were inflated with low-melting point agarose, and, after cooling, cut into ∼180 µm-thick sections as previously described [24]. Thin-cut lung slices (TCLSs) were mounted in a perfusion chamber on an inverted microscope and exposed to increasing concentrations of OxPAPC at room temperature. Video images were captured for 3 min, and acute airway narrowing calculated as a per cent decrease from the initial lumen volume. Exposure to PSPC or methacholine was used as negative and positive controls, respectively.
Statistics
We used a sparse partial least squares discriminant analysis (sPLS-DA, mixOmics 6.10.4, www.mixomics.org) to identify OxPCs important for distinguishing subjects with AHR from those without AHR or allergen response from diluent response. The optimal number of OxPCs required to discriminate each group was determined through an iterative tuning process in the sPLS-DA algorithm. OxPCs selected from the sPLS-DA analysis were then correlated with the clinical parameters of each subject using a Spearman's correlation matrix in R (corrplot version 0.8.4; R Foundation for Statistical Computing, Vienna, Austria). Spearman's correlation was used because the data did not fit a normal distribution. TCLS data were fit with a non-linear dose-response curve (Graphpad Prism 8, www.graphpad.com/scientific-software/prism/) and the half maximal effective concentration (EC50) calculated from the curve. An unpaired t-test was used to compare the maximal airway narrowing induced by OxPC 80 µg·mL−1 to the narrowing induced by PSPC at the equivalent concentration. All other data were analysed using a one-way ANOVA with a Dunnet's post hoc comparison. Kinome differential phosphorylation results were calculated using the Platform for Integrated, Intelligent Kinome Analysis 2 software [25]. Significantly upregulated phospho-signals were analysed using kinase enrichment analysis [26] to identify underlying activated kinases in the kinome data. The significance threshold was set at p<0.05. Experiments were completed in at least triplicate. Data analysis was done in R (version 3.5.1) and graphs made using ggplot2 package (version 3.1.1) or superheat (version 0.1.0).
Results
OxPCs in the DE3 cohort
PLS-DA analysis was able to cluster individuals with or without AHR separately based on their OxPC profiles (figure 1a). Using sPLS-DA, we were able to identify the most significant discriminative OxPCs of each group (figure 1b). There were eight OxPCs that discriminated the AHR group and five that discriminated the no-AHR group. PGPC and KODA-PPC were the most important discriminators of the two groups (highest loading weights, figure 1b). PGPC had the highest mean value in the group without AHR, and KODA-PPC had the highest mean value in the group with AHR. There was no correlation between sex and weight with any of the eight OxPCs that discriminated groups. There was a significant negative correlation between subject age and the abundance of PLPC-OH (supplementary figure S1). To identify the relationships between OxPCs identified by sPLS-DA and the degree of AHR, we performed a correlation analysis. For the full cohort, the slope of the methacholine dose-response curve (dose-response slope) was positively correlated with HDiA-PC (R=0.63) and KODA-PPC (R=0.64) abundance, and negatively correlated with PGPC abundance (R= −0.87) (p<0.05, figure 1c–e). Furthermore, the provocative concentration causing a 20% fall in forced expiratory volume in 1 s (FEV1) (PC20) of methacholine negatively correlated with SGPC abundance (R= −0.6, p<0.05, figure 1f) and positively correlated with PGPC abundance (supplementary figure S1). Finally, FEV1 positively correlated with PGPC abundance, and FEV1/forced vital capacity (FVC) ratio positively correlated with PGPC and PLPC-keto abundance (supplementary figure S1). A correlation matrix for all clinical measures listed in table 1 is shown in supplementary figure S1.

Oxidised phospholipid profiles differ in the lung dependent on airway hyperresponsiveness (AHR). a) Partial least squares discriminant analysis (PLS-DA) discriminated between individuals with and without AHR in the DE3 cohort based on oxidised phosphatidylcholine (OxPC) profile. b) 13 OxPC species were identified using sparse PLS-DA analysis as significant discriminators of individuals with and without AHR. The x-axis is a relative scale indicating the importance of each OxPC (higher absolute number is more important) in each group (AHR and no AHR). c) Correlation between HDiA-PC, d) KODA-PPC and e) PGPC in bronchoalveolar lavage fluid (BALF) with the methacholine dose-response slope (an index of methacholine responsiveness). f) Correlation between SGPC and the provocative concentration of methacholine causing a 20% fall in forced expiratory volume in 1 s (methacholine PC20). Data are plotted on a log scale. n=8 AHR and n=5 no AHR.
OxPCs in the DC cohort
PLS-DA analysis segregated and clustered response to allergen challenge from diluent challenge in individuals based on their OxPC profile (figure 2a). Using sPLS-DA analysis, we identified seven OxPCs in BALF that were associated with the allergen challenge. Seven different BALF OxPCs emerged as the most significant discriminating components for each group (figure 2b). KDiA-PC was the most important OxPC for separating the groups, and was most abundant in the BALF post-allergen challenge. The discriminating OxPCs significantly correlated with the severity of the late asthma response (percentage fall in FEV1). Specifically, PAPC-keto (R=0.69), PLPC-OH (R=0.66) and PLPC-OOH,OH (R=0.78) were all positively correlated with the percentage fall in FEV1 during the late asthma response (p<0.05, figure 2c–e). There was no clear association between the type of allergen used for inhaled challenge and the abundance of specific OxPCs. Furthermore, there was no association between sex or age and any of the selected OxPCs. A correlation matrix for all clinical measures listed in table 2 is shown in supplementary figure S2.

Oxidised phospholipid profiles change after allergen challenge in subjects with mild asthma. a) Partial least squares discriminant analysis (PLS-DA) discriminated allergen (blue) and diluent (orange) challenge in individuals with mild asthma based on their oxidised phosphatidylcholine (OxPC) profile. b) Seven OxPC species were identified using a sparse PLS-DA analysis as significant discriminators of subjects, post allergen or diluent challenge. The x-axis is a relative scale indicating the importance of each OxPC (higher absolute number is more important) in each group (allergen and diluent challenge). c) Correlation between PAPC-keto, d) PLPC-OH and e) PLPC-OOH,OH and the fall in forced expiratory volume in 1 s (FEV1) during the late asthma response. Data are plotted on a log scale. n=10 subjects with pre- and post-allergen bronchoalveolar lavage fluid (BALF). HDM: house dust mite.
OxPAPC effects on inflammatory cytokine production by HASM cells
To determine the effect of OxPAPC, a family of OxPCs that includes many identified in figures 1 and 2, we exposed HASM cells to OxPAPC over a range of concentrations that included levels we detected in BALF. This challenge induced secretion of interleukin (IL)-6, IL-8 and granulocyte–macrophage colony-stimulating factor (GM-CSF) (figure 3a–c). Specifically, OxPAPC 160 µg·mL−1 significantly increased IL-6 (349.9±200.3 pg·mL−1), IL-8 (107.2±95.8 pg·mL−1) and GM-CSF (67.3±52.2 pg·mL−1) abundance in cell culture media compared to the response to a non-oxidisable control phospholipid, PSPC (p<0.05). OxPAPC (80 µg·mL−1) also induced a significant accumulation of secreted IL-8 (104.8±61.5 pg·mL−1). Conversely, we detected no change in the abundance of interferon-γ (IFN-γ), IL-1β, IL-2, IL-4, IL-5, IL-10, IL-12(p70), IL-13, monocyte chemoattractant protein-1 (MCP-1) or tumor necrosis factor-α (TNF-α) in response to any concentration of OxPAPC.

Oxidised PAPC (OxPAPC) exposure of human airway smooth muscle (HASM) cells induces pro-inflammatory cytokine release. Abundance in cell culture media of a) interleukin (IL)-6, b) IL-8 and c) granulocyte–macrophage colony-stimulating factor (GM-CSF) in response to increasing concentrations of OxPAPC for 24 h (the numbers in x-axis labels indicate the concentration in μg·mL−1). Control indicates vehicle-only exposure. PSPC was used as a negative control at 160 μg·mL−1 (PSPC 160). d) Dose-dependent increase in cyclooxygenase-2 (COX2) mRNA abundance in response to OxPAPC. e) Temporal effect of 80 μg·mL−1 OxPAPC on COX2 mRNA abundance for up to 24 h exposure. f) Immunoblot showing the effects of OxPAPC on protein abundance for COX2 and COX1 in HASM cells. PSPC was used as negative control. NT: no treatment, vehicle-only control. Enhanced chemiluminescence (ECL) was required to detect COX2 protein in HASM cells treated with OxPAPC 80 μg·mL−1. *: p<0.05 determined by one-way ANOVA with Dunnett's post hoc test versus PSPC-treated cells. n=3 primary HASM cell cultures from different donors.
OxPAPC effects on COX2 and oxylipin production by HASM cells
Exposure of cells to OxPAPC resulted in a dose-dependent increase in COX2 mRNA abundance compared to PSPC-exposed HASM cells (figure 3d). Specifically, there was a significant elevation in COX2 mRNA abundance 24 h after exposure to 40 µg·mL−1 (10.5±7.5 fold increase), 80 µg·mL−1 (23.7±11.0 fold increase) and 160 µg·mL−1 (102.1±82.5 fold increase) of OxPAPC (p<0.05). COX2 mRNA abundance increased to a peak 1 h after OxPAPC exposure and remained elevated at least 24 h later (figure 3e). Induction of COX2 was confirmed by immunoblotting, showing a significant accumulation of COX2 protein at OxPAPC 160 µg·mL−1 compared to PSPC and no treatment (figure 3f). Exposure to OxPAPC 80 µg·mL−1 also resulted in accumulation of COX2 protein, but it was necessary to employ a more sensitive chemiluminescence reagent to reveal this response. There was no change in COX1 protein abundance in response to OxPAPC. Concomitantly, a significant dose-dependent accumulation of oxylipins, including prostaglandins and leukotrienes, was measured in response to 24 h exposure to OxPAPC (figure 4a). In total, we detected 72 different oxylipin species, and 32 were significantly increased by exposure to OxPAPC (p<0.05, figure 4a). A full table of all detected oxylipins is included in supplementary table S2. Figure 4 also shows the four oxylipins with the steepest dose-response relationship with different concentrations in the OxPAPC challenge. These included 6-trans leukotriene B4 (LTB4) (figure 4b), 12(S)-LTB4 (figure 4c), 5,15-dihydroxyeicosatetraenoic acid (5,15 diHETE) (figure 4d) and 12-hydroxyeicosatetraenoic acid (12-HETE) (figure 4e).

Oxidised PAPC (OxPAPC) exposure of human airway smooth muscle (HASM) cells significantly increases the production of oxylipin metabolites. A full list of detected oxylipins is provided in supplementary table S2. a) Heatmap showing the profile of oxylipin production in cell culture media of three different primary HASM cell cultures after 24 h exposure to increasing doses of OxPAPC (labelled on the x-axis as OxPC with the concentration in μg·mL−1). As a negative control, cells were challenged with PSPC 80 μg·mL−1. NT: no treatment (vehicle-only control); ns: nonsignificant. Each column represents a single cell culture. The 72 individual oxylipins are represented in each row (labelled on left). Abundance is demonstrated by colour from yellow to red (low abundance to high abundance), using z score (−2 minimum, +4 maximum). b) 6-trans leukotriene B4 (6-trans LTB4), c) 12(S)-LTB4, d) 5,15-dihydroxyeicosatetraenoic acid (5,15 diHETE) and e) 12-hydroxyeicosatetraenoic acid (12-HETE) had the most significant concentration-dependent relationships in response to OxPAPC exposure. *: p<0.05 one-way ANOVA with Dunnett's post hoc test versus PSPC treatment (PS160). n=3 primary HASM cell cultures from different donors.
OxPAPC induces airway narrowing
We found that OxPCs associate with AHR in human subjects. Therefore, we tested whether OxPAPC was sufficient to induce airway narrowing in murine TCLS. We found dose-dependent airway narrowing with OxPAPC, with an EC50 of 29.5 µg·mL−1 and a maximum closure of 15.4%±2.5% (figure 5). This was significantly different from PSPC, which failed to induce any airway narrowing. As a positive control we also measured methacholine dose-response curves, which revealed an EC50 of 250 nM, and indicated that maximum airway narrowing achieved by OxPAPC was equivalent to that induced by 34.4 nM of methacholine.
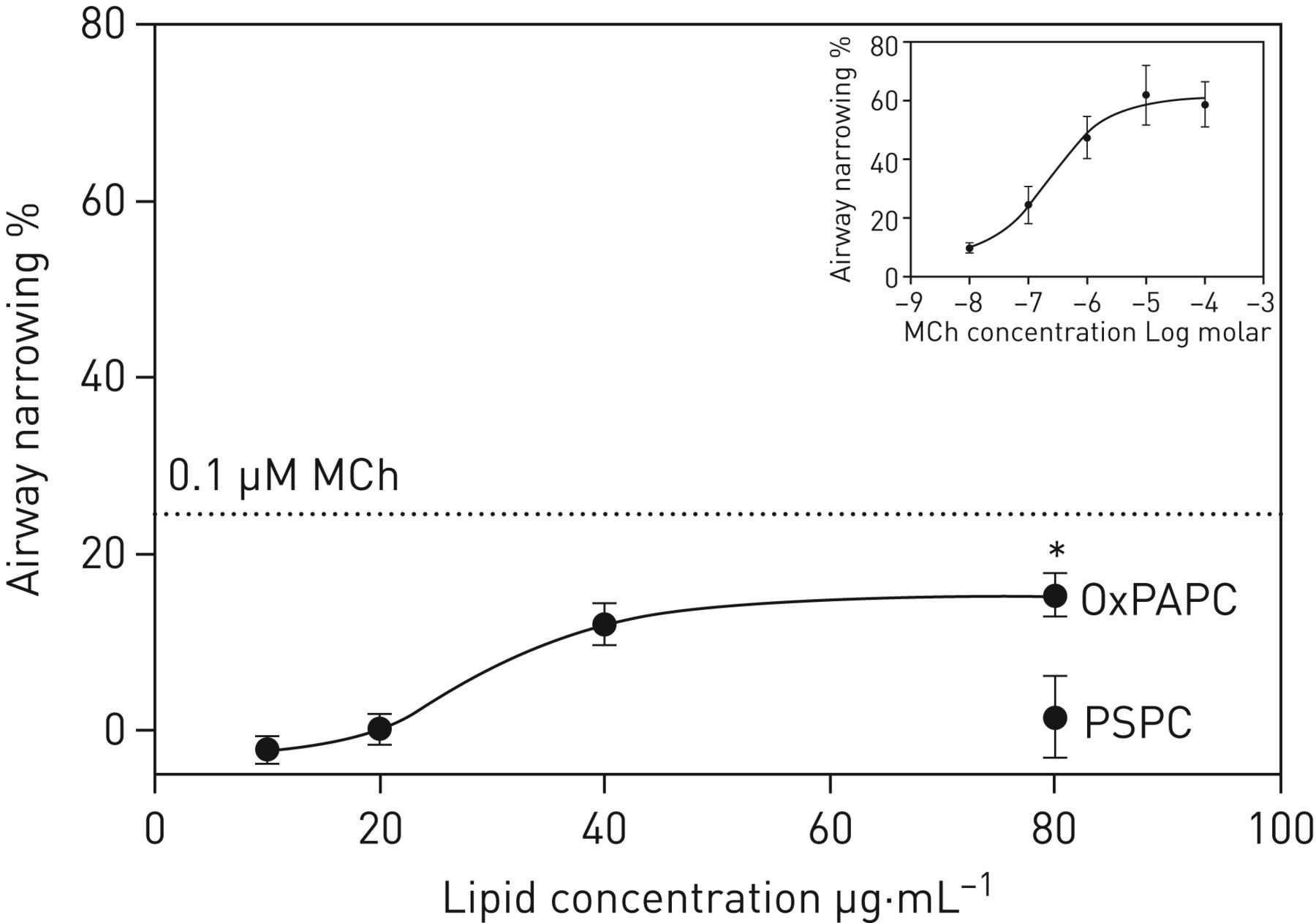
Oxidised PAPC (OxPAPC) exposure causes dose-dependent airway narrowing in murine thin-cut lung slices (TCLS). Dose–response curve comparing the effect of increasing concentration of OxPAPC on airway narrowing in a TCLS. *: p<0.05 versus PSPC 80 μg·mL−1. Dashed line indicates airway narrowing caused by 0.1 μM methacholine (MCh). Insert shows a reference MCh dose–response curve. Half maximal effective concentration for OxPAPC was 29.5 μg·mL−1 and for MCh was 0.24 μM.
OxPAPC signalling networks in HASM cells
In an attempt to decipher the receptors that may mediate the HASM cell response to OxPAPC, we screened a number of small molecular inhibitors, based on published predicted targets for OxPCs [27]. Figure 6a shows that inhibitors of the prostaglandin E2 receptors EP1 or EP2, Toll like receptors TLR2 and TLR4, platelet-activating factor receptor, or the macrophage scavenger receptor CD36 had no impact on OxPAPC-induced COX2 mRNA accumulation. To gain insight into the signalling networks that are activated in HASM cells in response to OxPAPC exposure, we performed a kinome analysis 1 h, 3 h and 6 h after treatment with OxPAPC (table 3). Given that 80 µg·mL−1 OxPAPC falls within the predicted physiological range of OxPCs in BALF after allergen challenge, we chose this concentration for kinomic and inhibitor studies. The phospho-signature that emerged revealed that the activity of 15 kinases was enriched after 1 h exposure (p<0.05), and the activity of 20 kinases was enriched after 3 h and 6 h OxPAPC exposure (p<0.05). Only two kinases activated at 1 h remained so through 6 h: pyruvate dehydrogenase kinase 1 (PDK1) and protein kinase Cδ (PRKCD).

Receptors and signalling pathways that regulate oxidised PAPC (OxPAPC)-induced cyclooxygenase 2 (COX2), oxylipins, cytokines and chemokines in primary human airway smooth muscle (HASM) cells. a) Relative abundance of OxPAPC-induced COX2 mRNA following 2-h pre-incubation with inhibitors for prostaglandin E2 receptor (PF-04418948 1 µM), prostaglandin E1/2 receptor (AH-6089 10 µM), TLR2/4 receptor (sparstolonin B 10 µM), platelet-activating factor receptor (WEB-2086 10 µM) or CD36 receptor (sulfosuccinimidyl oleate 20 µM). b) Relative abundance of OxPAPC-induced COX2 mRNA following 2-h pre-incubation with inhibitors for protein kinase C (GF-109203X 10 µM) or ROCK1/2 (Y-27632 10 µM). c) Interleukin (IL)-6, d) IL-8 and e) granulocyte–macrophage colony-stimulating factor (GM-CSF) abundance following 2-h pre-inhibition with GF-109203X, indomethacin (10 µM) or AH-6089 prior to the addition of OxPAPC. NT: no treatment. *: p<0.05 one-way ANOVA with Dunnett's post hoc test versus OxPAPC 80 µg·mL−1. n=3 primary HASM cell cultures from different donors.
Kinase enrichment results following 1, 3 or 6 h OxPAPC exposure in human airway smooth muscle cells
To validate the role of these predictions in the biological response to OxPAPC, we assessed the effect of protein kinase C (PKC) inhibition using GF-109203X. We targeted PKC because it is a confirmed signalling hub for OxPAPC effects on human endothelial cells [28]. As a negative control we assessed the inhibitory effects of Y-27632 on Rho-associated coiled-coil containing protein kinase (ROCK) 1 and ROCK2, which were not identified by our kinase enrichment array. PKC inhibition, but not ROCK1/2 inhibition, significantly decreased OxPAPC-induced COX2 mRNA accumulation by 64.8%±16.3% (p<0.05) (figure 6b). Moreover, PKC inhibition decreased OxPAPC-induced IL-6, IL-8 and GM-CSF by 87.5%±3.8%, 97.0%±1.9% and 98.6%±0.2%, respectively (p<0.05, figure 6c–e). To assess the potential confounding effects of COX2 metabolites to induce inflammatory cytokines biogenesis, we inhibited either COX2 activity using indomethacin, or prostaglandin signalling via E1/2 receptors using AH-6089. Indomethacin selectively prevented OxPAPC-induced GM-CSF production by 81.8%±9.0%. Similarly, inhibition of prostaglandin receptor signalling with AH-6089 selectively reduced OxPAPC-induced GM-CSF production by 79.5%±2.7%. Indomethacin and AH-6089 had no effect on OxPAPC-induced IL-6 and IL-8 release.
Discussion
By combining data from human cohorts, murine TCLS and primary cultured airway cells, we have revealed the presence of bioactive oxidised phospholipids in the airways of people with asthma and AHR. We have demonstrated their role in promoting airway narrowing and inducing a pro-inflammatory phenotype in HASM cells. Inhalation challenge by clinically relevant aeroallergens in humans resulted in the accumulation of a profile of OxPCs. Several OxPC variants were associated with key features of asthma and AHR, including methacholine dose-response slope, PC20 and the severity of fall in FEV1 during the late asthma response to an inhaled allergen. These data, combined with the ability of OxPAPC to narrow murine airways ex vivo, suggest an interaction between OxPCs and airway cells such as airway smooth muscle that results in greater airway narrowing and responsiveness. For this reason, we validated a potential role for OxPCs in asthma pathobiology using HASM cells. We found that OxPCs promote the expression of COX2 and the release of pro-inflammatory mediators, including IL-6, IL-8, GM-CSF and a panel of oxylipins replete with prostaglandins, leukotrienes and epoxyeicosatrienoic acids. Using a novel kinome array approach, we have uncovered a complex signalling response to OxPCs wherein diverse kinases are temporally induced, suggesting sequential signalling activation to orchestrate broad biological responses. We have revealed sustained activation of PKC, and used a selective inhibitor to validate its role in OxPC-induced inflammatory mediator release by HASM cells. Collectively, our study has identified OxPCs as new mediators of allergic asthma pathobiology and pathophysiology, and provides direction for translational and clinical research of their role in existing needs in asthma management.
We have demonstrated that inhaled allergen challenge of subjects with atopic asthma induces airway OxPC accumulation that correlates with clinically relevant early and delayed airway constriction. This suggests that OxPCs can contribute directly to a bronchoconstrictor response, a scenario we validated with murine TCLS preparations, using OxPAPC at a concentration equivalent to that in BALF after allergen challenge to induce 15% airway narrowing. Poiseuille's equation predicts this could increase airflow resistance by as much as 40%, and thus be sufficient to account for the 20% drop in FEV1 observed in subjects after allergen challenge. OxPC accumulation in the airways is consistent with the compromised antioxidant systems in the lungs of asthmatic subjects [7], and that oxidative stress markers [29], including serum malondialdehyde (a product of OxPC formation [30]), are elevated in mild-moderate asthma [31]. The asthmatic subjects we investigated were not treated with inhaled corticosteroids prior to sample collection. In any case, salmeterol and fluticasone reportedly do not reduce serum malondialdehyde, or presumably OxPC formation, to levels found in healthy controls [31]. A novel aspect of our work is that we investigated non-asthmatic individuals with AHR (DE3 cohort) and found that OxPCs were higher than in non-AHR subjects and correlated with airway dysfunction. This indicates that increased levels of OxPCs may be a pre-disposing factor for airway dysfunction, even in subjects without a clinical diagnosis of asthma. The effects of OxPCs are complex, given that low concentrations can enhance endothelial barrier function [12, 32] and have anti-inflammatory effects [15, 33]. Thus, a low abundance of OxPAPC in the airways may have natural homeostatic benefit, but an elevation in OxPAPC levels, as we report here, is sufficient to promote airway hyperreactivity in asthmatic and non-asthmatic individuals.
OxPCs are associated with multiple pathogenic pathways linked to chronic human diseases. OxPAPC promotes atherosclerotic lesion development and endothelial dysfunction [34–40]. They are also exacerbating factors in acute lung injury [13, 41], renal ischaemia–reperfusion injury [12] and acute or chronic inflammatory pain [42]. Cigarette smoke exposure produces lung OxPAPC that impairs macrophage phagocytosis and bacterial clearance [43]. A general role for OxPCs in pathobiology likely lies in the formation of oxidation-specific epitopes that are recognised by soluble and cell-associated pathogen recognition receptors, such as TLR2/4/6, CD36, C-reactive protein and the complement system [44]. Although OxPAPC can interact with G-protein-coupled receptors (GPCRs), including prostaglandin receptors and platelet-activating factor receptor [45, 46], using selective inhibitors we were unable to identify a specific surface receptor that mediates effects in HASM cells. This suggests a complex interface that requires broad profiling of OxPAPC cell signalling cascades.
Using kinome array we identified a temporally dynamic panel of OxPAPC-induced kinases in HASM cells. This could reflect interactions with multiple receptors and direct effects on cell membranes [47]. OxPAPC had disparate activation effects on kinases, with some rapidly but only transiently induced, and others only active 6 h after exposure. This suggests feed-forward signalling stemming from an initial response, but it was not possible to decipher pathway integration with our current study design. Our primary objective here was, instead, to validate the biological relevance of kinome array findings. For this purpose we targeted PKC in subsequent studies for three reasons. First, PKC was activated rapidly and remained active for up to 6 h. Second, Birukov et al. [28] and others [48] show that PKC is an important signalling hub for OxPAPC in endothelial cells. Third, OxPCs may disrupt lipid rafts that localise GPCRs and Gαq/11 subunits that are directly upstream of PKC [49]. Future work is needed to unravel the role of individual OxPC subtypes, and the receptor- or non-receptor-mediated processes that they trigger. Based on our findings here, this could include the exploration of other kinases that are associated with asthma pathobiology, including tyrosine kinase 2 and janus kinase 1 [50], which was rapidly activated, and pyruvate dehydrogenase kinase 1 [51], which underwent sustained activation after OxPAPC exposure.
OxPAPC exerted a strong, but somewhat selective, pro-inflammatory response in HASM cells. This included the release of IL-6, IL-8 and GM-CSF, which are associated with eosinophilic and neutrophilic airway inflammation in allergic asthma [52–54], with IL-8 and GM-CSF also associated with severe asthma [55, 56]. This suggests OxPCs could be linked to severe, steroid-resistant asthma, a point that should be addressed in future. We further showed that PKC activation is necessary for OxPAPC induction of IL-6, IL-8 and GM-CSF. This confirms the biological significance of our kinome assay, and is consistent with multiple reports that PKC regulates cytokine transcription and secretion in smooth muscle and other airway cells [57–60]. The 13-plex panel used in this study included the relatively narrow array of asthma-associated cytokines produced by HASM cells, but a wider profile of mediators from in vivo asthma models is needed to fully establish how OxPCs may orchestrate airway inflammation and immunity.
Our experiments with HASM cells showed that OxPAPC, even at relatively low concentrations, induces oxylipin biosynthesis, including products that require COX2 activity. On this basis we specifically investigated the effects on COX2, and found that biologically relevant concentrations of OxPAPC were sufficient to increase COX2 mRNA and protein, though immunoblotting was not sensitive enough to detect changes induced by concentrations <80 μg·mL−1. OxPAPC exposure also increased the abundance of the 5-lipoxygenase product LTB4, which promotes inflammation and the late asthma response, and is elevated in severe asthma [61–64]. Furthermore, OxPAPC induced the production of 15/12-lipoxygenase products 5,15-diHETE and 12-HETE, which have less well-defined roles in asthma but have been implicated in modulating inflammation, with chemoattractant roles for eosinophils [65, 66]. Future research should focus on how OxPCs promote the production of these potent lipid mediators and their role in inflammation and AHR.
As presented in figure 7, we have uncovered the broad effects of OxPAPC on HASM cells, and suggest that processes for PKC activation are linked to lipid and protein mediator biosynthesis. Novel mechanisms for OxPC effects are likely, because even though COX2 and oxylipins are induced, pro-inflammatory mediators such as IL-1β or TNF that are typically associated with COX2 [67, 68] are not. We show that OxPC activation of PKC is needed to induce COX2, and this is consistent with responses to other stimuli [69, 70]. We also show that inhibiting COX2 activity or E1/E2 receptor-mediated response to COX2 metabolites blocks GM-CSF, but not IL-6 or IL-8 biosynthesis, whereas PKC inhibition effectively prevents release of all three cytokines. This diversity in pathway activation and integration is also evident in the kinome response to OxPC that we tracked in HASM cells. It appears that OxPAPC-induced oxylipins may potentiate cytokine release in a paracrine fashion, with the potential for positive feedback pathways to perpetuate COX2 expression [71]. Notably, we could not identify a receptor that directly mediates OxPC-induced COX2. Thus, feed-forward signalling may be critical, resulting from disruption of cell membranes, the creation of oxidation-specific epitopes and the formation of lipid and peptide products and cellular debris that can activate various receptors. Identifying these secondary products and their receptors is an important area for future study. In this regard, the requirement of PKC activity for perpetuating the effects of OxPCs suggests that Gq/11- and Gi-coupled GPCR agonists (e.g. prostaglandins, prostacyclins and thromboxane) and 5-lipoxygenase products (e.g. LTB4) may be candidates. Coincidently, many Gq/11 GPCRs for 5-lipoxygenase and COX2 products, and receptors for some cytokines, including GM-CSF, mediate mitogen-activated protein kinase activity, which is linked to sustained COX2 activation [72–74]. Collectively, our findings create the need for new research to identify receptor and non-receptor processes affected by OxPCs in HASM cells, other lung structural cells and immune cells in the context of allergic asthma and AHR.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic overview of signalling pathways orchestrating oxidised PAPC (OxPAPC)-induced cyclooxygenase 2 (COX2) and inflammatory cytokine synthesis. OxPAPC causes airway narrowing through an unidentified cell membrane receptor or direct effects on the plasma membrane. By similar receptor or membrane-linked mechanisms, OxPAPC also activates protein kinase C (PKC), which leads to production of interleukin (IL)-6 and IL-8 and expression of COX2. COX2 products (oxylipins) signal through the prostaglandin E2 receptor 1/2 (EP1/2) and are required for oxidised phosphatidylcholine (OxPC)-induced production of granulocyte–macrophage colony-stimulating factor (GM-CSF).
Our study design imposes some limitations on the interpretation of our data. Surprisingly, we found no significant change in the total OxPC abundance in our human samples, even after allergen challenge. The natural biological variability of individual human subjects may explain this, but it is also possible that the OxPC profile is the critical determinant of biologically significant OxPC production. This creates the need for more precise analysis of associations between clinical traits and the abundance of individual OxPCs in larger cohorts. Our study was not designed to directly compare broad differences in total OxPC abundance between subjects; rather, our goal was to highlight the presence of OxPCs in the human lung and to profile the association of differences in AHR and response to allergen challenge on the profile of OxPC species. For this reason, the subjects in the two cohorts we studied did not have severe airway disease and the subjects with diagnosed asthma (DC cohort) had well-controlled disease at the time of sample collection. To more effectively determine the influence of asthma severity, asthma treatment, environmental exposures and sex on OxPC profile and abundance, an investigation of sufficiently powered cohorts is needed. Nonetheless, our current work is the first that reveals an association, and testable biological mechanisms, for oxidised phospholipids in the pathophysiology of chronic airway disease.
In summary, we show for the first time that airway OxPCs are present in the airways and correlate with lung dysfunction in individuals with AHR and in subjects with mild asthma after allergen challenge. Our data using cultured HASM cells indicates that airway structural cells exhibit a coordinated secretory response involving lipid mediators, cytokines and chemokines. This discovery opens new avenues to explore the role of bioactive molecules linked with oxidative stress in airway disease, and suggests that OxPCs could represent one mechanism for persistent inflammation that requires new options for asthma management.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00839-2020_Supplementary_material
Figure S1 ERJ-00839-2020_Figure_S1
Figure S2 ERJ-00839-2020_Figure_S2
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00839-2020.Shareable
Footnotes
This article has an editorial commentary: https://doi.org/10.1183/13993003.03736-2020
This article has supplementary material available from erj.ersjournals.com
Author contributions: C.D. Pascoe performed cell culture experiments, data analysis and preparation of manuscript draft. A. Jha performed isolation of OxPC from human samples, data analysis and preparation of manuscript draft. M.H. Ryu performed isolation of OxPC for DE3 BAL sample and assisted in collecting samples for DE3 study. J. Vaghasiya performed and analysed studies using thin-cut lung slices and preparation of the manuscript. S. Basu assisted with animal experiments and editing of the manuscript. M. Ragheb and G.L. Stelmack contributed to collection of data from cell culture experiments and data analysis. S. Srinathan and B. Kidane recruited thoracic surgery subjects and collected human lung specimens that were used to generate primary HASM cell cultures, and they edited the manuscript. J. Kindrachuk performed the kinomics array on cell culture lysates. A. Ravandi contributed to experimental design and performed OxPC analyses in human samples. C. Carlsten, G.M. Gauvreau and P.M. O'Byrne performed human exposures and collected human samples. A.J. Halayko conceived the project, its design and data plan, and contributed to drafting the manuscript.
Conflict of interest: C.D. Pascoe has nothing to disclose.
Conflict of interest: A. Jha has nothing to disclose.
Conflict of interest: M.H. Ryu has nothing to disclose.
Conflict of interest: M. Ragheb has nothing to disclose.
Conflict of interest: J. Vaghasiya has nothing to disclose.
Conflict of interest: S. Basu has nothing to disclose.
Conflict of interest: G.L. Stelmack has nothing to disclose.
Conflict of interest: S. Srinathan has nothing to disclose.
Conflict of interest: B. Kidane has nothing to disclose.
Conflict of interest: J. Kindrachuk has nothing to disclose.
Conflict of interest: P.M. O'Byrne has received consulting and/or speakers fees from AstraZeneca, GSK, Chiesi and Meranari, and grants in aid from AstraZeneca, Medimmune, GSK, Novartis and Merck, outside the submitted work.
Conflict of interest: G.M. Gauvreau has nothing to disclose.
Conflict of interest: A. Ravandi has nothing to disclose.
Conflict of interest: C. Carlsten has nothing to disclose.
Conflict of interest: A.J. Halayko has nothing to disclose.
Support statement: Funding was received from CIHR-ICRH Emerging Network Grant for the Canadian Respiratory Research Network (CIHR (MOP 123319)), WorkSafe BC (RG2011-OG07) and AllerGen National Centre for Excellence (GxE4). Funding information for this article has been deposited with the Crossref Funder Registry. J. Kindrachuk is funded by a Tier 2 Canada Research Chair in the Molecular Pathogenesis of Emerging and Re-Emerging Viruses provided by the CIHR (Grant no. 950-231498). A.J. Halayko is funded by a Tier 1 Canada Research Chair in Lung Pathobiology and Treatment. C. Carlsten is funded by both Tier 2 Canada Research Chair and an AstraZeneca Chair in Occupational and Environmental Lung Disease. A. Ravandi is supported by the FW Du Val Clinical Research Professorship from the Manitoba Medical Services Foundation. C.D. Pascoe is supported by a CIHR Banting Fellowship. J. Vaghasiya is supported by a PhD Studentship from Research Manitoba and the Children's Hospital Research Institute of Manitoba.
- Received March 24, 2020.
- Accepted August 26, 2020.
- Copyright ©ERS 2021
References










