





Abstract
Background A randomised controlled trial in Japan showed that inhaled N-acetylcysteine monotherapy stabilised serial decline in forced vital capacity (FVC) in some patients with early idiopathic pulmonary fibrosis (IPF). However, the efficacy and tolerability of combination therapy with an antifibrotic agent and inhaled N-acetylcysteine are unknown.
Methods This 48-week, randomised, open-label, multicentre phase 3 trial compared the efficacy and tolerability of combination therapy with pirfenidone plus inhaled N-acetylcysteine 352.4 mg twice daily with the results for pirfenidone alone in patients with IPF. The primary end-point was annual rate of decline in FVC. Exploratory efficacy measurements included serial change in diffusing capacity of the lung for carbon monoxide (DLCO) and 6-min walk distance (6MWD), progression-free survival (PFS), incidence of acute exacerbation, and tolerability.
Results 81 patients were randomly assigned in a 1:1 ratio to receive pirfenidone plus inhaled N-acetylcysteine (n=41) or pirfenidone (n=40). The 48-week rate of change in FVC was −300 mL and −123 mL, respectively (difference −178 mL, 95% CI −324–−31 mL; p=0.018). Serial change in DLCO, 6MWD, PFS and incidence of acute exacerbation did not significantly differ between the two groups. The incidence of adverse events (n=19 (55.9%) for pirfenidone plus N-acetylcysteine; n=18 (50%) for pirfenidone alone) was similar between groups.
Conclusions Combination treatment with inhaled N-acetylcysteine and pirfenidone is likely to result in worse outcomes for IPF.
Abstract
We compared the efficacy of pirfenidone plus inhaled N-acetylcysteine with results for pirfenidone alone for IPF. Combination treatment with inhaled N-acetylcysteine and pirfenidone is likely to result in worse outcomes for IPF. https://bit.ly/3eWEbvW
Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating, progressive and fatal disorder characterised by worsening dyspnoea and progressive loss of lung function [1]. The only currently approved evidence-based treatments recommended in international IPF guidelines are pirfenidone and nintedanib [2], which have been licenced and used to treat IPF worldwide, e.g. in European and Asian countries and the United States.
Pirfenidone exhibited anti-inflammatory, antioxidant and antifibrotic effects in experimental models of pulmonary fibrosis, and thus has therapeutic potential for IPF in humans [3–6]. Pirfenidone was better than placebo in preserving forced vital capacity (FVC) and improving progression-free survival (PFS) in patients with IPF [7, 8]. Results from the phase 3 CAPACITY [9] and ASCEND [10] trials showed that pirfenidone significantly slowed disease progression and might improve survival.
Several other unapproved medications are used to treat IPF, including N-acetylcysteine, which is a tripeptide (γ-glutamyl-cysteinyl-glycine), a precursor of the antioxidant glutathione (GSH) and a scavenger of oxygen free radicals. N-acetylcysteine directly alters the structure of transforming growth factor-β and thus attenuates its putative profibrotic properties [11]. Oxidant–antioxidant imbalance may contribute to IPF disease progression [12–17]. N-acetylcysteine is a potentially effective therapeutic compound for IPF, as it may replenish GSH stores, thereby restoring the natural oxidant–antioxidant balance and preventing oxidative injury preceding fibroproliferation [16, 17].
In addition to the antifibrotic effect of pirfenidone, N-acetylcysteine and pirfenidone act at different points in reactive oxygen species (ROS) metabolism. N-acetylcysteine, together with catalase, is believed to decompose hydrogen peroxide into oxygen and water. In contrast, pirfenidone exerts its beneficial effects on ROS metabolism by scavenging toxic hydroxyl radicals [3–6]. High-concentration aerosol administration of N-acetylcysteine acts directly as an antioxidant in alveoli [15, 16]. Thus, there is a logical basis for combining them to greatly reduce ROS.
Although oral N-acetylcysteine has been studied in numerous clinical trials, the results have been discouraging when it is given as a single agent or in combination with pirfenidone to patients with IPF. Data from clinical trials of oral N-acetylcysteine for IPF include the results of the PANTHER trial, in which a 60-week regimen of oral N-acetylcysteine 600 mg three times daily was not superior to a matched placebo in maintaining FVC in patients with IPF [18, 19].
The PANORAMA study assessed the safety and tolerability of oral N-acetylcysteine (600 mg daily) combined with pirfenidone in patients with IPF. Data on change in FVC indicated that the addition of oral N-acetylcysteine to pirfenidone was unlikely to improve outcomes. Indeed, the decrease in FVC was greater for combination therapy than for pirfenidone alone [20]. Oral N-acetylcysteine may not yield a sustained increase in GSH levels that is sufficient to increase the antioxidant capacity of the lungs, even when given in high doses [21, 22]. However, in addition to increasing GSH, aerosol administration of N-acetylcysteine acts directly as an antioxidant in alveoli [15, 16]. N-acetylcysteine inhalation might therefore reduce inflammation and lung fibrosis. Although a randomised trial of inhaled N-acetylcysteine monotherapy did not achieve the primary end-point in all participants, post hoc analysis showed a significantly lower 48-week decline in FVC in a subset of patients [23]. Furthermore, in a small Japanese case–control study of 27 patients with progressive disease, outcomes were better for inhaled N-acetylcysteine and pirfenidone than for pirfenidone alone [24].
The updated international IPF guidelines conditionally recommend against the use of oral or inhaled N-acetylcysteine monotherapy for treatment of IPF [2]. The guidelines state that further research is required in order to determine whether some subgroups of patients might benefit from N-acetylcysteine monotherapy. However, little evidence indicates that N-acetylcysteine monotherapy is effective for IPF. In addition, because of the lack of evidence from randomised controlled trials, no recommendation was given regarding the combined use of inhaled N-acetylcysteine with an antifibrotic agent [2].
The efficacy and tolerability of combination therapy with an antifibrotic agent and inhaled N-acetylcysteine are unclear. Therefore, this study compared the efficacy and safety of combination treatment with pirfenidone and inhaled N-acetylcysteine 352.4 mg twice daily with the results for pirfenidone alone for treatment of IPF over a 48-week treatment period.
Methods
Study design and participants
In this phase 3, randomised, open-label study, patients aged >40 years with a diagnosis of IPF were recruited at 50 medical centres in Japan. IPF was diagnosed in accordance with the 2011 IPF international guidelines [1]. All adults with a confirmed clinical and radiological diagnosis of IPF were eligible for inclusion.
High-resolution computed tomography (HRCT) scans were reviewed by a central reading committee, to verify eligibility according to the protocol and serial change in HRCT images (images were obtained within 1 month of study inclusion and after 24 weeks and 48 weeks of treatment). Patients were included if they had an FVC >50% and a diffusing capacity of the lungs for carbon monoxide (DLCO) >35% and had been receiving pirfenidone 1200–1800 mg·day−1 before randomisation.
Patients with severe or unstable concomitant disease were excluded from the study. During the study, patients were not permitted to receive any treatment for IPF other than the study medication (inhaled N-acetylcysteine) and pirfenidone. Concomitant treatment with up to 20 mg of prednisolone per day, or the equivalent, was permitted if the dose had been stable for ≥8 weeks before screening; patients receiving other treatments for IPF, including nintedanib, high-dose prednisolone, immunomodulators or any investigational treatment for IPF were excluded. If acute exacerbation was reported by an investigator at any time during the trial, any treatment could be initiated, or the dose increased, as deemed appropriate by the investigator. In addition, patients were excluded if the results of physiological tests or HRCT scanning were better than those recorded 6 months previously. Patients with coexisting pulmonary arterial hypertension, bronchial asthma, sarcoidosis, bronchiectasis, neoplasm or infectious disease were also excluded.
Study treatment
Patients received either pirfenidone plus inhaled N-acetylcysteine or pirfenidone alone. Inhaled N-acetylcysteine therapy was administered by using Micro Air nebulisers and vibration mesh technology (NE-U07; Omron, Tokyo, Japan). Patients receiving N-acetylcysteine were treated twice daily with 352.4 mg of N-acetylcysteine, which was diluted with saline to a total volume of 6–8 mL.
Pirfenidone (Shionogi & Co., Osaka, Japan) was continued throughout the 48-week study period and was administered orally three times daily with food. Patients were expected to receive 1200–1800 mg pirfenidone per day. Dose interruption or a dose reduction from 1800 mg to 1200 mg was allowed for management of adverse events. After an adverse event had resolved, the dose was increased to 1800 mg.
Patients were enrolled by the principal investigator at each site and randomised at a 1:1 ratio to receive pirfenidone plus N-acetylcysteine or pirfenidone alone. Randomisation was performed with the use of a minimisation method, and the study drug was assigned by using an interactive internet-response system. After the 48-week treatment period, patients reported for a follow-up visit, 4 weeks later. Spirometry was conducted at baseline, at 12, 24, 36 and 48 weeks, and at the follow-up visit. To minimise the number of missing data, patients who prematurely discontinued the study drug were asked to attend all scheduled visits and undergo all originally planned examinations.
End-points
The primary end-point was annual rate of decline in FVC (measured in mL·year−1). Key secondary end-points were PFS (progression was defined as a 10% decline in FVC from baseline, development of acute exacerbation or death) and incidence of acute exacerbation (as reported by site investigators) assessed over the 48-week treatment period [1, 10, 25]. The data were collected at baseline and at each study visit (at weeks 12, 24, 36 and 48).
Other pre-specified secondary end-points included absolute change from baseline in FVC (in millilitres and as a percentage of the predicted value) and diffusing capacity (DLCO) over the 48-week treatment period, the proportion of patients with an FVC response (defined as the proportion of patients in whom the percentage of predicted FVC declined by >5%, at weeks 24 and 48), 6-min walk distance (6MWD), change from baseline in the COPD Assessment Test (CAT) score [26] and modified Medical Research Council (mMRC) dyspnoea score over the 48-week treatment period, and death from any cause. All mortality end-points were recorded as the time to death. Additionally, changes in serum levels of markers of interstitial pneumonia (Krebs von den lungen (KL)-6 and surfactant protein (SP)-D) during the 48-week treatment period were evaluated.
Safety was assessed by means of clinical and laboratory evaluations at study visits and the recording of adverse events. Acute exacerbations were defined as events that satisfied all the following criteria: unexplained worsening or development of dyspnoea within the previous 30 days; new diffuse pulmonary infiltrates visualised on chest radiography and/or HRCT or development of parenchymal abnormalities with no pneumothorax or pleural effusion (new ground-glass opacities) since the preceding visit; and exclusion of any known causes of acute worsening, including infection, left heart failure, pulmonary embolism and any identifiable cause of acute lung injury, in accordance with routine clinical practice and microbiological studies.
Statistical analysis
Analyses were done on the intention-to-treat population, which included all patients who were randomly assigned and received at least one dose of study medication (pirfenidone plus N-acetylcysteine or pirfenidone alone). All values are expressed as mean±sd, and differences between patient groups were analysed using the Wilcoxon test, t-test and Fisher exact test for two independent samples.
Changes from baseline values were compared using the Wilcoxon test. Categorical variables were compared using the Fisher exact test and continuous variables were compared using the Wilcoxon test. Incidence rates were analysed using the Fisher exact test. For missing values, the principle of last observation carried forward was used. Analyses of change in FVC from baseline were performed using ANCOVA using the respective baseline measurements as covariates. ANCOVA was used to compare changes in other pulmonary function test results and serum levels of markers of interstitial pneumonia. For time-to-event analyses, the log-rank test was used to compare treatment groups. All reported p-values are two-sided and were considered statistically significant when <0.05. All analyses were performed using SAS software (version 9.4; SAS Institute, Cary, NC, USA). Safety was assessed by means of clinical and laboratory evaluations at study visits and the recording of adverse events.
The sample size was calculated to provide 85% power to detect a between-group difference of 90 mL in the annual rate of FVC decline. On the basis of data from a phase 3 Japanese trial of pirfenidone [8], the standard deviation for change in FVC from baseline was assumed to be 160 mL in both groups. Assuming that it would not be possible to evaluate data for 25% of patients, the sample size was calculated as 75 patients in the pirfenidone group and 75 patients in the pirfenidone plus N-acetylcysteine group for a two-group t-test with a one-sided significance level of 2.5%.
Ethics
The study was performed in accordance with the International Conference on Harmonisation Guidelines, the Declaration of Helsinki and with the relevant local legal and regulatory requirements applicable to the countries in which the trial was conducted. This study was conducted with the approval of the institutional review board of Toho University Omori Medical Center (project approval number 26-174). Approval from institutional review boards or institutional ethics committees at each study site was obtained before the start of the study. Written informed consent was obtained from patients before screening. A data monitoring committee of independent experts was responsible for reviewing emerging adverse events throughout the trial.
Results
Patient characteristics
Because of unexpectedly slow enrolment, this clinical trial was completed after 81 of the planned 150 subjects were enrolled. The first patient entered the study on June 22, 2015, and the last patient completed the 48 weeks of treatment on June 29, 2018. 41 patients were randomly assigned to treatment with pirfenidone plus N-acetylcysteine, and 40 were randomly assigned to treatment with pirfenidone. Seven patients in the pirfenidone plus N-acetylcysteine group and nine patients in the pirfenidone group did not complete treatment and were excluded from the data analysis. Baseline characteristics, including lung function, were generally well balanced between treatment groups; mean percent predicted FVC values were similar in the pirfenidone plus N-acetylcysteine and pirfenidone groups. Median (range) duration from IPF diagnosis was 42 (2–158) months in the pirfenidone plus N-acetylcysteine group and 36 (5–166) months in the pirfenidone group. Duration of pirfenidone use before randomisation was similar between the pirfenidone plus N-acetylcysteine group and pirfenidone group (255±515 versus 282±371 days; p=0.80). The proportion of patients using glucocorticoids at randomisation did not differ between the pirfenidone plus N-acetylcysteine group and pirfenidone group (0 (0%) versus 3 (8.3%); p=0.24). There was no difference in the extent or severity of the honeycomb pattern on HRCT. Analysis of HRCT findings in relation to IPF classification, as detailed in the 2011 international guideline, indicated a definite usual interstitial pneumonia (UIP) pattern in 80.5% of patients and possible UIP pattern in 19.5% of patients in the pirfenidone plus N-acetylcysteine group, and a definite UIP pattern in 70.0% of patients and possible UIP pattern in 30% of patients in the pirfenidone group (table 1). Of the 70 patients who received the study drug during the 48-week treatment period, 34 in the pirfenidone plus N-acetylcysteine group and 36 in the pirfenidone group completed the study (figure 1). All patients received pirfenidone before randomisation.
Summary of baseline demographic characteristics

Study flowchart. PI: principal investigator.
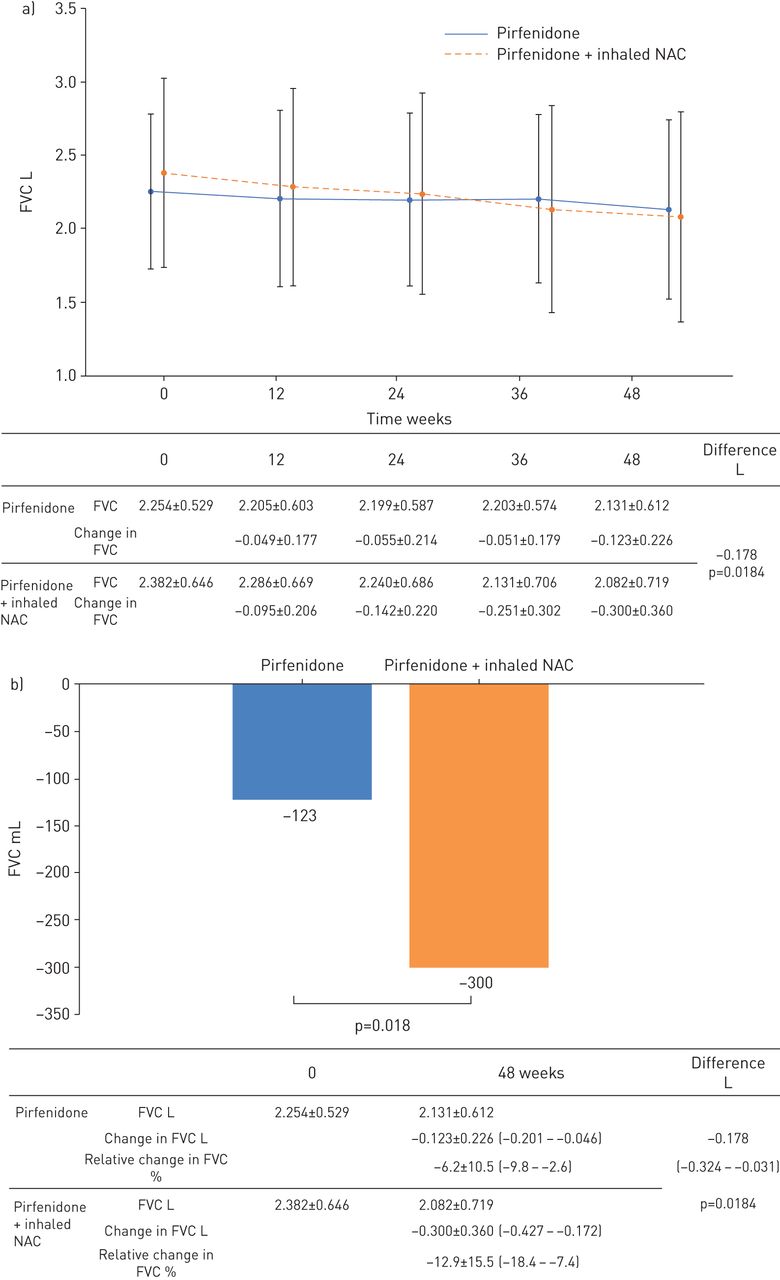
Serial change from baseline in FVC volume is shown in figure 2a. The 48-week change in FVC was −300 mL in the pirfenidone plus N-acetylcysteine group and −123 mL in the pirfenidone group (difference −178 mL, 95% CI −324–−31; p=0.018) (figure 2b). The 48-week change in FVC values indicated that the addition of N-acetylcysteine to pirfenidone was unlikely to yield clinical benefits and might instead worsen outcomes in patients with IPF. In the per-protocol population, change in FVC exhibited a similar trend (−311 mL in the pirfenidone plus N-acetylcysteine group and −160 mL in the pirfenidone group). The standard effect size for change in FVC was 0.583 (95% CI 0.097–1.069). The upper bound of the 95% confidence interval of the effect size is 1.069; thus, any positive effect of N-acetylcysteine is likely to be small. Change in FVC was −8.38% predicted in the pirfenidone plus N-acetylcysteine group and −4.35% pred in the pirfenidone group (difference −4.20, 95% CI 8.44–0.04; p=0.052) (table 2). Differences were observed between the pirfenidone plus N-acetylcysteine and pirfenidone groups in the proportion of patients with ≥5% decline in FVC (64.7% (22 out of 34) versus 44.4% (16 out of 36), respectively) and the proportion of patients with improved/unchanged FVC (35.3% (12 out of 34) versus 52.8% (19 out of 36), respectively) at week 48. Change in percent predicted DLCO was similar between groups (−10.13% pred versus −7.37% pred, respectively) at week 48 (table 2).

The 48-week change in forced vital capacity (FVC). a) FVC before and after 48 weeks of pirfenidone administration in the inhaled pirfenidone plus N-acetylcysteine group and pirfenidone monotherapy group. b) The 48-week change in FVC was significantly greater in the pirfenidone plus N-acetylcysteine group than in the pirfenidone monotherapy group (−300 mL versus −123 mL, respectively; difference −178 mL, 95% CI −324–−31 mL; p=0.018).
Secondary lung function end-points at week 48
There was no significant difference in PFS between the pirfenidone plus N-acetylcysteine and the pirfenidone group (48.1 versus 49.6 weeks, respectively; p=0.94) (figure 3) or incidence of acute exacerbation (one (2.9%) out of 34 versus four (11.1%) out of 36; p=0.185). Mean change in 6MWD from baseline to week 48 was −78.1 m in the pirfenidone plus N-acetylcysteine group and −24.2 m in the pirfenidone group (p=0.252) (table 2). The proportion of patients with worse disease at 48 weeks, as comprehensively assessed by HRCT imaging, was 39.3% in the pirfenidone plus N-acetylcysteine group and 21.4% in the pirfenidone group (p=0.257). Serum markers of pneumocyte injury, including KL-6 and SP-D, did not differ significantly between the two groups. CAT scores were similar between the two treatment groups at week 48. Mean change from baseline in CAT score at week 48 was +3.8 in the pirfenidone plus N-acetylcysteine group and +1.2 in the pirfenidone group. mMRC was similar between the two treatment groups during the 48-week treatment period.

{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier curves for progression-free survival (PFS) in the combination therapy group (n=34) and pirfenidone monotherapy group (n=36). PFS did not differ significantly between the treatment groups (p=0.94). NAC: N-acetylcysteine.
Safety
The incidence of adverse events (n=19 (55.9%) receiving pirfenidone plus N-acetylcysteine; n=18 (50.0%) receiving pirfenidone alone) was similar between groups. 10 serious adverse events were reported, but there was no significant difference in the rate of such events in the pirfenidone plus N-acetylcysteine (four (11.8%) out of 34) and pirfenidone (six (16.7%) out of 36) groups (table 3). Five deaths occurred during the study, but none was considered related to treatment: three patients in the pirfenidone alone group died from worsening IPF, including acute exacerbation, and one patient in the pirfenidone plus inhaled N-acetylcysteine group died from pulmonary haemorrhage due to fungal infection.
Summary of adverse events
One (2.8%) patient in the pirfenidone group and three (8.8%) patients in the pirfenidone plus N-acetylcysteine group developed photosensitivity, but improved after treatment with steroid ointment and ultraviolet ray care and did not withdraw. 10 patients developed gastrointestinal symptoms such as anorexia and nausea, but the rates of such symptoms did not differ between the two groups.
The incidence of bacterial pneumonia was not significantly different between the present groups, and no serious adverse events were attributed to inhaled N-acetylcysteine. Therefore, addition of inhaled N-acetylcysteine to pirfenidone does not substantially alter pirfenidone tolerability.
Discussion
In contrast to previous findings from a case–control study of inhaled N-acetylcysteine in combination with pirfenidone [24], FVC data from this prospective open-label randomised trial suggest that inhaled N-acetylcysteine has no clinical benefit when combined with pirfenidone for treatment of IPF. In fact, decline in FVC over 48 weeks was greater for patients treated with pirfenidone plus N-acetylcysteine than for those treated with pirfenidone alone, which indicates that, as compared with pirfenidone alone, combination therapy did not improve the decline in FVC.
In the PANORAMA study, data on change in FVC indicated that addition of oral N-acetylcysteine to pirfenidone was unlikely to be of clinical benefit [20]. Similarly, our study suggests that combination therapy did not further slow the decline in FVC, as compared to pirfenidone alone. In addition, inhaled N-acetylcysteine may not be advantageous to oral N-acetylcysteine, despite the higher concentration in the lungs. Although PFS and incidence of acute exacerbation were similar between treatment groups, most secondary lung function end-points and HRCT findings at week 48 also tended to be worse in the pirfenidone plus N-acetylcysteine group. These results suggest that inhaled N-acetylcysteine is not beneficial in IPF when combined with pirfenidone and that the combination might result in worse outcomes for patients with IPF. None of the evaluated end-points was better in the pirfenidone plus N-acetylcysteine group.
A post hoc analysis of data from the PANTHER trial suggested that polymorphisms in the gene encoding Toll-interacting protein (TOLLIP; rs3750920) modify the response to oral N-acetylcysteine. The authors hypothesised that N-acetylcysteine could be beneficial for patients with the T/T genotype, but may be associated with worse outcomes in patients with the C/C genotype, possibly by reducing defence against infection and thereby hastening IPF progression. These results suggest a potential mechanism that can explain why pirfenidone provides a benefit with N-acetylcysteine when administered to patients with a T/T TOLLIP genotype, but cannot prevent the harmful effect of N-acetylcysteine in patients with the C/C genotype, which results in greater decline in FVC [27]. Koyama et al. [28] investigated polymorphisms in the TOLLIP genotype in Japanese IPF patients and reported a very low frequency of the T/T genotype, for which N-acetylcysteine may be beneficial. In contrast, the frequency of the C/C genotype (for which N-acetylcysteine might be harmful) was high. Thus, genotype frequency may have influenced the present results. The present patients did not provide samples for genotype analysis; therefore, we were unable to confirm this hypothesis in patients receiving N-acetylcysteine combination treatment. Present and previous results indicate that we should not use off-label N-acetylcysteine or conduct a new clinical trial without genetic analysis before patient selection.
A previous study found that bacterial pneumonia, cough and sore throat were common adverse events of inhaled N-acetylcysteine [23]. In this randomised, open-label, controlled trial, the combination of inhaled N-acetylcysteine and pirfenidone 1200–1800 mg·day−1 was generally well tolerated in patients with IPF. The safety profile of the inhaled N-acetylcysteine and pirfenidone combination was similar to that of pirfenidone alone, which differs from the findings of a case–control study of inhaled N-acetylcysteine and pirfenidone compared with pirfenidone. The PANORAMA study [20] reported greater photosensitivity in the pirfenidone plus oral N-acetylcysteine combination group; however, photosensitivity did not differ significantly in the present study: photosensitivity was noted in one patient in the pirfenidone group and in three patients in the inhaled N-acetylcysteine combination group.
All adverse events in the previous study were likely caused by pirfenidone and were reported in the group treated with inhaled N-acetylcysteine and pirfenidone. Our data do not suggest that inhaled N-acetylcysteine affects pirfenidone tolerability. Concomitant administration of inhaled NAC was generally well tolerated.
This study has several limitations. First, this is an open-label study, not a placebo-controlled study, and the design might have resulted in bias that affected outcomes. Second, all patients in this study are Japanese, and racial differences may need to be considered when evaluating the effectiveness of the drug. Third, we could not confirm adherence to inhaled N-acetylcysteine treatment. Finally, although the planned sample size was 150, only 81 patients were randomised because of slow enrolment. This smaller sample size limits the conclusions that can be drawn from the data.
In conclusion, our analysis of the primary end-point shows that decline in FVC was greater in patients treated with pirfenidone plus inhaled N-acetylcysteine than in those treated with pirfenidone alone. Thus, inhaled N-acetylcysteine combined with pirfenidone may result in worse outcomes for patients with IPF. The present results indicate that inhaled N-acetylcysteine, as administered in this trial, should not be used to treat IPF.
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00348-2020.Shareable
Footnotes
This article has an editorial commentary: https://doi.org/10.1183/13993003.03551-2020
This phase 3 trial was registered with the University Hospital Medical Information Network (UMIN) (registration number: UMIN000015508). No provision has been made for individual participant data to be publicly available
Conflict of interest: S. Sakamoto has nothing to disclose.
Conflict of interest: K. Kataoka has nothing to disclose.
Conflict of interest: Y. Kondoh reports advisory board fees and personal fees from Asahi Kasei Pharma Corp., Boehringer Ingelheim Co. Ltd and Shionogi & Co. Ltd; advisory board fees from Janssen Pharmaceutical K.K; and personal fees from Eisai Inc., Kyorin Pharmaceutical Co. Ltd, Mitsubishi Tanabe Pharma and Novartis Pharma K.K, outside the submitted work.
Conflict of interest: M. Kato has nothing to disclose.
Conflict of interest: M. Okamoto has nothing to disclose.
Conflict of interest: H. Mukae reports grants and personal fees from Shionogi & Co, Ltd, during the conduct of the study.
Conflict of interest: M. Bando reports personal fees from Shionogi & Co, Ltd, outside the submitted work.
Conflict of interest: T. Suda has nothing to disclose.
Conflict of interest: K. Yatera reports grants from Kirigaoka Tsuda Hospital, Tochiku Hospital, Hagiwara Central Hospital, Kurate Hospital, Saiseikai Yamaguchi Hospital, Ono Pharmaceutical Co. Ltd, Teijin Home Healthcare Limited, Taiho Pharmaceutical Co. Ltd, Daiichi Sankyo Company, Limited, GlaxoSmithKline K.K, Pfizer Japan Inc., Taisho Pharma Co. Ltd, MSD K.K, Novartis Pharma K.K, Nippon Boehringer Ingelheim Co. Ltd, Daiwa Securities Health Foundation, Actelion Pharmaceuticals Japan Ltd, Astellas Pharma Inc., AstraZeneca K.K, Eisai Co., Ltd, Shionogi & Co., Ltd, KYORIN Pharmaceutical Co. Ltd, Taisho Pharma Co. Ltd, Daiichi Sankyo Company, Limited, Sumitomo Dainippon Pharma Co. Ltd, Chugai Pharmaceutical Co. Ltd, Teijin Pharma Limited, and Eli Lilly Japan K.K, outside the submitted work.
Conflict of interest: Y. Tanino has nothing to disclose.
Conflict of interest: T. Kishaba has nothing to disclose.
Conflict of interest: N. Hattori has nothing to disclose.
Conflict of interest: Y. Taguchi has nothing to disclose.
Conflict of interest: T. Saito has nothing to disclose.
Conflict of interest: Y. Nishioka reports grants and personal fees from Shionogi & Co. Ltd, during the conduct of the study; and grants and personal fees from Nippon Boehringer Ingelheim Co. Ltd, MSD K.K, Ono Pharmaceutical Co. Ltd, Taiho Pharmaceutical Co. Ltd, Chugai Pharmaceutical Co. Ltd, Asahi Kasei Pharma Corporation and Eli Lilly Japan K.K, and grants from Bonac Corporation, outside the submitted work.
Conflict of interest: K. Kuwano has nothing to disclose.
Conflict of interest: K. Kishi reports personal fees from Shionogi, outside the submitted work.
Conflict of interest: N. Inase has nothing to disclose.
Conflict of interest: S. Sasaki has nothing to disclose.
Conflict of interest: H. Takizawa has nothing to disclose.
Conflict of interest: T. Johkoh has nothing to disclose.
Conflict of interest: F. Sakai has nothing to disclose.
Conflict of interest: S. Homma has nothing to disclose.
Support statement: This study was supported by research grants from the Ministry of Health, Labour and Welfare of Japan and the study group for strategic exploration of drug seeds for Diffuse Lung Disease and construction of clinical evidence of the Japan Agency for Medical Research and Development (AMED) (15ek0109064h0002 and 16ek0109064h0003). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received February 18, 2020.
- Accepted July 16, 2020.
- Copyright ©ERS 2021
References










