





Abstract
DNA sequencing of the SERPINA1 gene to detect α1-antitrypsin (AAT) deficiency (AATD) may provide a better appreciation of the individual and cumulative impact of genetic variants on AAT serum levels and COPD phenotypes.
AAT serum level and DNA sequencing of the coding regions of SERPINA1 were performed in 1359 participants of the Canadian Cohort Obstructive Lung Disease (CanCOLD) study. Clinical assessment for COPD included questionnaires, pulmonary function testing and computed tomography (CT) imaging. Phenotypes were tested for association with SERPINA1 genotypes collated into four groups: normal (MM), mild (MS and MI), intermediate (heterozygote MZ, non-S/non-Z/non-I, compound IS, and homozygote SS) and severe (ZZ and SZ) deficiency. Smoking strata and MZ-only analyses were also performed.
34 genetic variants were identified including 25 missense mutations. Overall, 8.1% of alleles in this Canadian cohort were deficient and 15.5% of 1359 individuals were carriers of at least one deficient allele. Four AATD subjects were identified and had statistically lower diffusion capacity and greater CT-based emphysema. No COPD phenotypes were associated with mild and intermediate AATD in the overall cohort or stratified by smoking status. MZ heterozygotes had similar CT-based emphysema, but lowered diffusion capacity compared with normal and mild deficiency.
In this Canadian population-based cohort, comprehensive genetic testing for AATD reveals a variety of deficient alleles affecting 15.5% of subjects. COPD phenotype was demonstrated in severe deficiency and MZ heterozygotes. This study shows the feasibility of implementing a diagnostic test for AATD using DNA sequencing in a large cohort.
Abstract
15.5% of subjects in this Canadian cohort were carriers of at least one deficient allele affecting alpha-1 antitrypsin serum levels, but only genotypes resulting in severe deficiency and MZ heterozygotes were associated with COPD phenotypes https://bit.ly/3ekozCf
Introduction
α1-Antitrypsin deficiency (AATD) is an inherited disorder associated with accelerated rate of lung function decline and early onset emphysema [1–4]. AATD is caused by genetic mutations in the SERPINA1 gene located on chromosome 14, which encodes an antiprotease called α1-antitrypsin (AAT). The prevalence of AATD is estimated at one in 2000 to 5000 in North American populations [5, 6]. Previous studies suggest that severe AATD accounts for 1% to 5% of COPD cases [1, 7, 8], but the exact proportion for the COPD population remains to be described.
Diagnostic methods for AATD vary by country and region, but usually follow a multi-step testing algorithm that includes quantification of AAT in serum or plasma, protease inhibitor phenotyping by isoelectric focussing, and targeted genotyping for the most common mutations (e.g. S and Z) [9–11]. The gold standard to detect the deficiency is direct DNA sequencing, which was historically considered laborious, expensive and not available in all centres [2]. In the current genomic era, these arguments are no longer valid. More research and clinical laboratories are transitioning to DNA sequencing as the method of choice [11–15]. In so doing, there is an increasing number of rare variants being identified; however, little is known about their frequencies and clinical impact.
The goals of this study were two-fold: first, to assess the frequencies of AATD alleles in a Canadian population-based cohort of individuals with COPD, at risk of COPD, and never-smokers free of airway obstruction; and, secondly, to evaluate the individual and cumulative impact of AATD alleles on COPD phenotypes including lung function and computed tomography (CT)-based emphysema.
Methods
CanCOLD study
The Canadian Cohort of Obstructive Lung Disease (CanCOLD) is a prospective cohort study built on the Canadian COPD prevalence study “COLD”, which evaluated >5000 subjects (male and female subjects aged ≥40 years) recruited through a random sampling frame from nine urban and suburban areas in Canada (ClinicalTrials.gov identifier: NCT00920348) [16]. Sampling for CanCOLD consists of all COPD subjects as well as age- and sex-matched non-COPD peers (postbronchodilator forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) >0.70) from the COLD study. CanCOLD thus comprises two balanced COPD subpopulations (mild and moderate–severe) and two matched non-COPD subpopulations including ever-smokers (for those at risk) and never-smokers (for the control subjects). Assessments included sociodemographic and clinical status questionnaires (including the COPD Assessment Test, the St. George's Respiratory Questionnaire for COPD (SGRQ-C), mMRC dyspnoea scale and Short Form-36), full pulmonary function tests, CT imaging of the thorax and incremental maximal cardiopulmonary exercise testing. Details about pulmonary function testing and CT imaging are in the supplementary material. Detailed descriptions of the sampling strategy and assessments can be found in the published protocol [16]. A written informed consent was obtained from all subjects and the study was approved by the Institutional Research Ethical Board of each site. For this analysis, only data from the initial visit were used and a final set of 1359 CanCOLD subjects with DNA available were selected for SERPINA1 sequencing.
DNA sequencing of SERPINA1
The DNA sequences of the coding regions (i.e. exons 2 to 5) of the SERPINA1 gene were obtained by Sanger sequencing in the same laboratory for all subjects. For identified variants with unknown allelic background, allele-specific PCR (AS-PCR) was performed in order to amplify and sequence each allele independently. Details are in the supplementary material.
In silico functional analysis of genetic variants
The predicting damaging effects of coding non-synonymous variants were evaluated with PolyPhen-2 [17] and Sorting Intolerant From Tolerant (SIFT) [18]. The deleteriousness of identified genetic variants was also evaluated with the Combined Annotation–Dependent Depletion (CADD) framework [19]. Finally, clinical interpretation was queried in ClinVar [20].
Measurement of AAT serum levels
Blood samples from CanCOLD participants were drawn by venipuncture and processed within 2 h of collection. Serum aliquots were stored at −80°C until analysis. AAT serum levels were measured by immunoturbidimetry on a COBAS INTEGRA 800 analyser (Roche Diagnostics, Laval, QC, Canada). The coefficient of variation for intermediate precision was estimated at 2.8% at a level of 0.76 g·L−1 and 2.2% at a level of 1.79 g·L−1.
Statistical analysis
The effects of genetic variants on AAT serum levels were evaluated using Wald tests for quantitative phenotype as implemented in PLINK [21]. Clinical data were compared across groups using ANOVA for continuous variables and chi-square for categorical variables. Analyses were also carried out stratified by smoking status (never- and ever-smokers). Analyses were performed with and without adjustment for covariates including age, sex and study sites. Post hoc Tukey multiple pairwise-comparison tests were used for statistically significant ANOVA to identify groups that differed.
Results
Identified genetic variants in CanCOLD
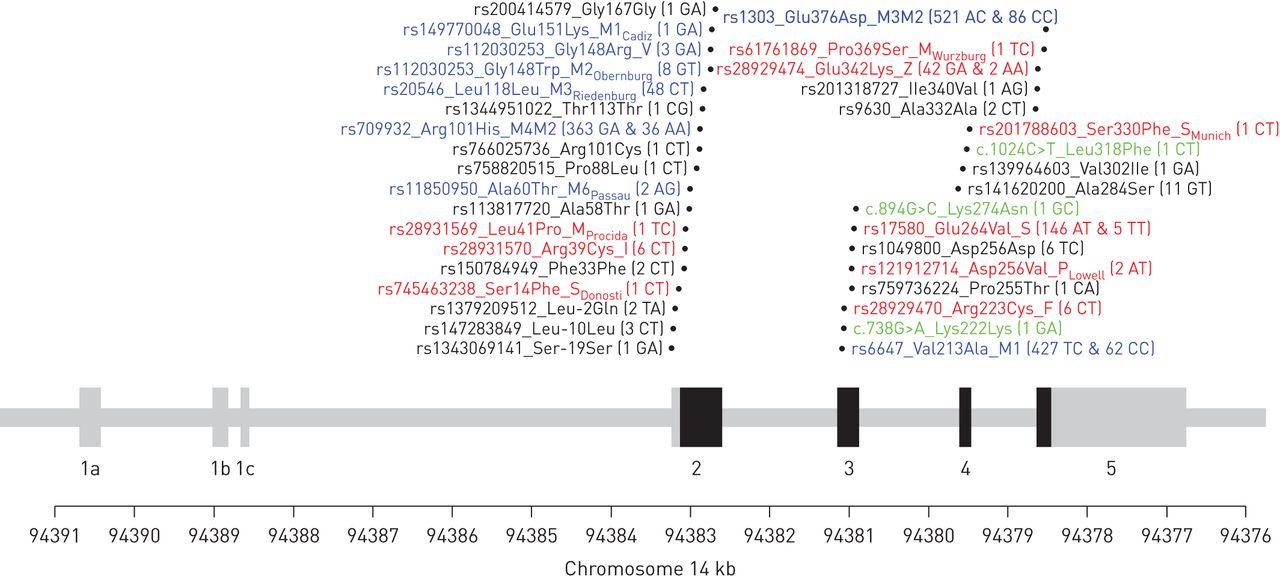
Table S1 shows the clinical characteristics of the 1359 CanCOLD participants evaluated in this study. DNA sequencing of exons 2 to 5 in these individuals identified 34 genetic variants. The location of these variants relative to the intron–exon structure of the SERPINA1 gene is illustrated in figure 1. Table 1 shows the variants based on standardised genetic, nucleotide, protein and protease inhibitor typing nomenclatures. For each variant, the pathogenicity scores and the allele frequency among the 1359 CanCOLD subjects are also provided in table 1. Three new variants were found including two in exon 3 (Lys222Lys and Lys274Asn) and one in exon 4 (Leu318Phe) (figure 1). These new variants are rare with allele frequencies of 0.04% (one heterozygote subject) and predict benign or inconsistent across pathogenicity scores (table 1).

The exon–intron structure of the SERPINA1 gene and the localisation of the identified genetic variants. The coding exons are shown in black and the untranslated regions in grey. Genetic variants are illustrated with their rs numbers (if available), protein nomenclature, and genotyping counts in parentheses for 1359 individuals. Newly identified variants are illustrated in green and named based on standard gene mutation nomenclature [27]. Variants causing a change in conventional protease inhibitor typing are illustrated in blue and red for normal and deficient alleles, respectively. Note that rs112030253 in exon 2 is indicated twice as it is a multi-allelic polymorphism and three alleles are observed in the CanCOLD cohort.
Nomenclature of the 34 genetic variants identified, allele frequencies in CanCOLD, and pathogenicity scores
Allele and genotype calling
DNA sequencing provides more granularity compared with the conventional protease inhibitor system. We thus had to use a refined strategy to call alleles and genotypes. Humans are diploid organism, meaning that there are two alleles per individual. For each allele, one or more genetic variants can be present. The absence or presence of a genetic variant is called in relation to an allele (DNA sequence) of reference. For SERPINA1, the allele of reference and the most common in human is labelled M1 (Val213) using the protease inhibitor system. In CanCOLD, for example, 1246 out of 2718 alleles (1359 individuals×2 alleles=2718 alleles) are on this background allele. Accordingly, we called the 2718 alleles and the 1359 genotypes of CanCOLD participants based on the absence or presence of the 34 genetic variants identified (figure 1 and table 1). Figure 2 illustrates five representative individuals to better conceptualise the allele and genotype calling process.

Schematic examples of allele and genotype calling derived from DNA sequencing data for five individuals. Case 1 is homozygote for the M1 (Val213) allele. This is the most common and normal allele in humans. The DNA nucleotide sequence of M1 is thus considered the reference. Case 2 is a typical MZ individual with one normal M1 allele and one deficient Z allele. The Z allele is the result of a missense mutation (Glu342Lys) in exon 5. Except for very rare cases, the Z allele is found on the M1 (Ala213) background. Case 3 is a SZ individual with one Z allele and one S allele characterised by the Glu264Val mutation in exon 3. Case 4 is carrier of the rare mutation Arg101Cys on the S background. Arg101Cys is considered deleterious based on pathogenicity scores (table 1). There are thus two hits (Cys101 and Val264) on the same allele. For this individual, knowing the allelic background was clinically relevant to determine the AATD status; i.e. the individual is heterozygote for one deficient allele and not compound heterozygote (two deficient alleles). Case 5 is carrier of a typical M3 allele characterised by Glu376Asp and a second unusual allele with His101 and Ala213, labelled R for rare allele background. These are all considered normal genetic variants, so there is no clinical implication of determining the allelic background of this individual. However, the observation is of great interest for population genetics. Note that this allele and genotype calling process was used to derive all alleles (n=2718) and genotypes (n=1359) of the 1359 CanCOLD individual.
Frequency of alleles
The 34 genetic variants in CanCOLD results in 40 distinct alleles. Table 2 shows the distribution of alleles in the overall CanCOLD cohort and by recruitment sites. The most frequent normal alleles in this population are 45.8% for M1 (Val213), 17.5% for M1 (Ala213), 14.6% for M2, 9.1% for M3, 1.8% for M3Riedenburg, and 1.3% for M4. In total, 91.9% of the 2718 alleles are normal. In contrast, the others are pathogenic, including known deficient or dysfunctional alleles such as S (5.7%), Z (1.7%), F (0.2%), I (0.2%) as well as more rare variants such as PLowell found in two heterozygote subjects and MProcida and MWurzburg each found in one heterozygote subject. Four additional rare variants, each found in a single individual, are claimed pathogenic by all scores (table 1) including M3.Pro255Thr, SDonosti, SMunich and S.Arg101Cys. Taking together, 220 deficient alleles are identified and indicate that 8.1% of alleles (220 out of 2718) in this Canadian cohort are deficient. The percentage of deficient alleles varies from 5.5% to 11% across recruitment sites (table 2).
Allele distribution sorted by allele frequency in the overall CanCOLD cohort and by recruitment sites sorted by geographic location from west to east
Genotype frequencies
The genotype of each individual consists of the combination of both alleles. Figure 3 illustrates the number of individuals in any combinations of normal and deficient alleles. Two subjects carry two copies of the Z allele and are thus confirmed cases of AATD. One other subject was compound heterozygote SZ. A second SZ individual was identified, but carrying also the Ser14Phe mutation. Allele-specific PCR indicated that this mutation occurs on the background of S allele, which is consistent with a previously reported allele labelled SDonosti [14]. As a result, there were a total of four CanCOLD subjects with severe AATD. 40 additional individuals were heterozygotes for the Z allele. Five SS individuals were found, and 143 additional individuals carried the S allele (142 MS and 1 IS). Six subjects were carriers of the F allele. Six carriers of allele I were also found including one IS. Rarer deficient alleles were identified in two PLowell, one MProcida, one MWurzburg, one SMunich and one M3.Pro255Thr heterozygote carriers. Overall, 210 out of 1359 subjects (15.5%) were carriers of at least one deficient allele.

Number of patients identified in any pairwise combinations of normal and deficient alleles. Alleles are annotated based on the legacy nomenclature, i.e. protease inhibitor system, if available, or on protein nomenclature. Normal alleles without protease inhibitor naming were combined with their respective background to ease visualisation (for example, allele M1.Val302Ile was merged with M1, this allele does not have a protease inhibitor naming and Val302Ile is benign). Each individual is represented in only one cell and the combinations of alleles for the 1359 CanCOLD participants are indicated. The colour of the squares illustrates the expected level of deficiency on a black and white scale where white indicates no deficiency and black indicates severe deficiency. *: Rare allelic background (1. His101 and Ala213 on the same allele, and 2. His101, Ala213, and Val340 on the same allele).
SERPINA1 genotyping grouping
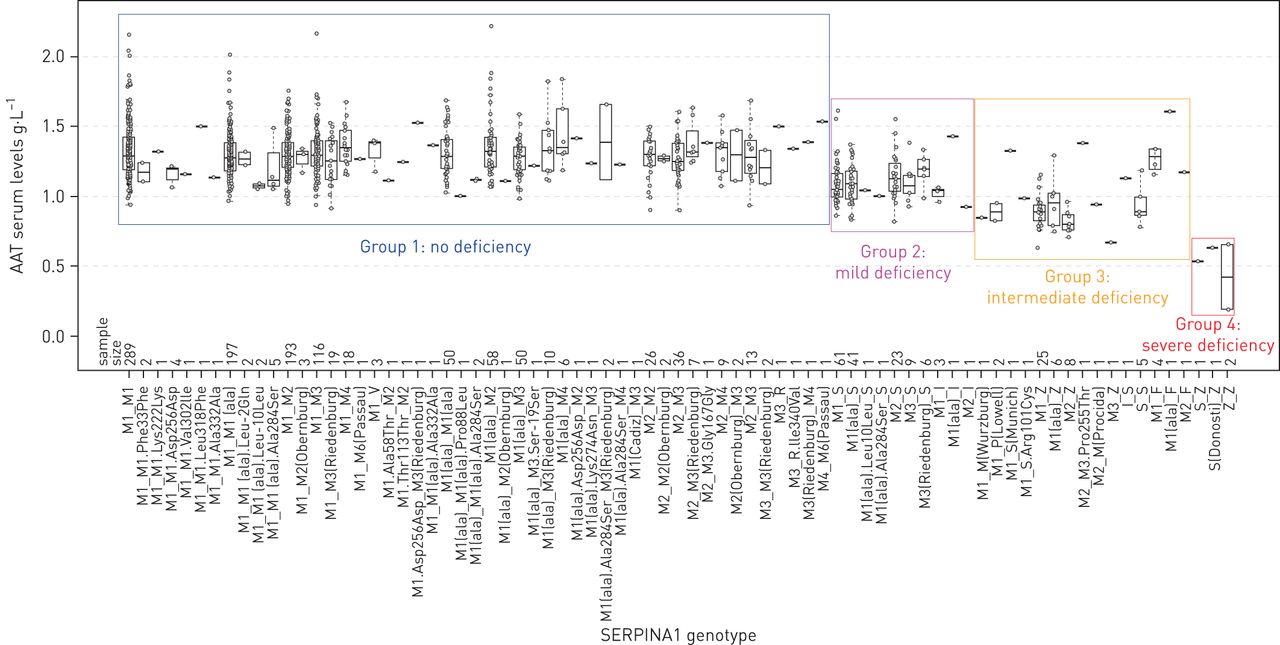
SERPINA1 genotyping groups were then generated from the DNA sequencing data to reflect the resulting AAT serum level or functional activity of AAT (figure 4). At the most detailed level, the 40 distinct alleles found in CanCOLD lead to 77 distinct genotypes with frequencies ranging from 0.07% (one out of 1359 individuals) to 21.3% (289 out of 1359 individuals, i.e. genotype M1M1) (see abscissa of figure 4). Although there is no standard for grouping genotypes, we have classified individuals into four groups based on the information of both alleles and the known or predicting effect of each genetic variant on AAT levels or activity. Group 1 (no deficiency) includes all individuals with two normal alleles (n=1149). Group 2 (mild deficiency) are individuals with one normal allele and one S or I allele (n=147, MS and MI). Group 3 (intermediate deficiency) consists of individuals that are heterozygotes (MZ, MF, MMProcida, MMWurzburg, MPLowell, MSMunich, MS.Arg101Cys, MM3.Pro255Thr), homozygotes for the S allele (SS) and compound heterozygotes for deficient alleles (IS) (n=59). Group 4 (severe deficiency) are AATD individuals (n=4, ZZ, SZ, and SDonostiZ).

Serum levels of α1-antitrypsin (AAT) by SERPINA1 genotypes. SERPINA1 genotyping groups are depicted by rectangles. Groups were generated to reflect the resulting impact of the genetic variants on AAT serum level or functional activity of AAT. For rare mutations of unknown biological and clinical significance, the grouping was guided based on the actual serum level of AAT among carriers and pathogenicity scores. This classification system results in four groups: Group 1 (no deficiency) includes all individuals with two normal alleles (n=1149). Group 2 (mild deficiency) are individuals with one normal allele and one S or I allele (n=147, MS and MI). Group 3 (intermediate deficiency) consists of individuals that are heterozygotes (MZ, MF, MMProcida, MMWurzburg, MPLowell, MSMunich, MS.Arg101Cys, MM3.Pro255Thr), homozygotes for the S allele (SS) and compound heterozygotes for deficient alleles (IS) (n=59). Group 4 (severe deficiency) are AATD individuals (n=4, ZZ, SZ and SDonostiZ). AAT serum levels are not available for 15 out of 1359 individuals including the single individual carrying the M1Cadiz allele, which explains the absence of boxplot for the M1(Cadiz)_M3 genotype. M3_R, “R” indicates rare allelic background.
Association of SERPINA1 genotyping groups with AAT serum levels and COPD phenotypes
Table 3 shows the clinical characteristics of CanCOLD participants according to the SERPINA1 genotyping groups. Serum AAT levels were strongly associated with genotyping groups (ANOVA p<0.001; figure 5a). The 6-heterozygote individuals for the F allele have serum AAT levels ranging with those with two normal alleles. Diffusion capacity to carbon monoxide (DLCO) and the percentage of lung voxels below −950 Hounsfield units (LAA−950) were also statistically different across these groups (table 3 and figure 5b and c). These associations were driven by AATD individuals (group 4: severe deficiency) as no significant differences were observed between group 1 (no deficiency) and group 3 (intermediate deficiency). No statistically significant differences were observed across these genotyping groups for smoking habits, lung function, symptoms and quality of life, self-reported comorbidities and respiratory medications (table 3).
Clinical characteristics by SERPINA1 genotyping groups

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serum levels of α1-antitrypsin (AAT) (a), diffusing capacity of the lung for carbon monoxide (DLCO) (b), and percentage of lung voxels below −950 Hounsfield units (LAA−950) (c) by SERPINA1 genotyping groups derived from DNA sequencing data. Genotyping groups are on the abscissa. Group 1 includes all individuals with two normal alleles (n=1149). Group 2 are individuals with one normal allele and one S or I allele (n=147, MS and MI). Group 3 consists of individuals that are heterozygotes (MZ, MF, MMProcida, MMWurzburg, MPLowell, MSMunich, MS.Arg101Cys, MM3.Pro255Thr), homozygotes for the S allele (SS) and compound heterozygotes for deficient alleles (IS) (n=59). Group 4 are AAT deficient individuals (n=4, ZZ, SZ and SDonostiZ). Boxplot boundaries represent the first and third quartiles, whiskers are the most extreme data points which are no more than 1.5-times the interquartile range, and the centre mark represents the median. Red dots illustrate the six individuals carrying one F allele. F allele have dysfunctional proteins but normal AAT blood levels [28], which exemplifies the intrinsic limitation of relying solely on serum measurement of AAT and commercially available genotyping tests to establish the diagnosis of AAT deficiency.
Intermediate AAT deficiency, mostly defined by MZ heterozygotes, was previously associated with an increased risk of COPD, especially in the presence of smoking exposure [22–25]. We thus repeated the analyses by smoking status (never- and ever-smokers). As observed for the entire cohort, the SERPINA1 genotyping groups were significantly different for LAA−950 in both never- and ever-smokers, and DLCO in never-smokers (table S2). Again, post hoc tests revealed that group 4 (severe deficiency), but not group 3 (intermediate deficiency), is significantly different from group 1 (no deficiency). We have also evaluated the interaction between SERPINA1 genotyping groups and smoking (ever versus never smokers) on COPD phenotypes, but no significant interactions were identified (table 3).
Analysis of MZ heterozygotes
Finally, COPD phenotypes were compared by SERPINA1 genotyping groups, but this time, limiting intermediate deficiency to MZ heterozygotes (n=40). Compared to normal and mild deficiency, MZ had similar lung function and CT-based emphysema (Table S3). However, DLCO was statistically lower (p<0.001) in MZ (95.5%±21.2%) and severe deficiency (69.4%±19.1%) compared with normal (106.4%±24.3%) and mild deficiency (109.9%±26.5%).
Discussion
In this Canadian cohort, 34 genetic variants were identified by sequencing the coding regions of the SERPINA1 gene. This includes genetic variants accounting for common normal alleles, such as M1, M1 (Ala213), M2, M3 and M4, but also more rare normal alleles, such as M3Riedenburg, M2Obernburg, V, M6Passau and M1Cadiz. Collectively, 91.9% of all the alleles evaluated were considered normal. In contrast, a variety of 220 deficient alleles was found in these 1359 individuals. Not surprisingly, the most common were S and Z, but mutations causing alleles I, F, PLowell, MProcida, MWurzburg, M3.Pro255Thr, SDonosti, SMunich and S.Arg101Cys were also identified. Deficient alleles were observed in 210 individuals, indicating that 15.5% of CanCOLD subjects carried at least one deficient allele. Grouping subjects by genotypes reflecting the serum level or functional activity of AAT indicated that severe deficiency (ZZ, SZ and SDonostiZ) had lower diffusion capacity and greater CT-based emphysema compared with subjects with no deficiency (MM). These differences were not observed in mild (MS and MI) and intermediate (heterozygote MZ, heterozygote non-S/non-Z/non-I, homozygote SS and compound heterozygote IS) deficiency. However, in MZ-only analysis, MZ heterozygotes had lower diffusion capacity compared with normal and mild deficiency.
Four subjects (0.29%) had AATD in CanCOLD, i.e. ZZ, SZ or SDonostiZ. This is lower than the 1.9% of ZZ found in 965 severe COPD patients [7], but higher than the frequency of severe AATD commonly reported at one out of 2000–5000 individuals [5, 6]. Random sampling in this study [16] which includes subgroups without airway obstruction, those at-risk of COPD, and individuals with COPD with a range of severity may explain this difference. Nevertheless, deficient alleles together including Z, S, I, F, PLowell, MProcida, MWurzburg, M3.Pro255Thr, SDonosti, SMunich and S.Arg101Cys account for 8.1% of all alleles tested (220 out of 2718). Overall, these alleles affect 15.5% of individuals, which may have a genetic predisposition to develop COPD [22, 23]. Our results highlight that less frequent deficient alleles, beyond S and Z, affects 20 individuals (1.5%) in CanCOLD.
In this study, we identified 59 individuals with intermediate AATD. The novelty of our study is that we have included genotypes associated with intermediate deficiency beyond MZ including heterozygote non-Z (MF, MMProcida, MMWurzburg, MPLowell, MSMunich, MS.Arg101Cys, MM3.Pro255Thr), homozygote SS, and compound heterozygote IS. However, the COPD phenotypes of this group was not statistically different compared to individuals with no deficiency (MM), which was consistent across smoking status strata. Sample size may have limited our ability to detect the effect of intermediate deficiency on COPD phenotypes. Alternatively, the ascertainment method based on random sampling in CanCOLD may also explain this lack of association. For many COPD cohorts, ascertainment is based on patients referred specifically for obstructive airway disease. In the natural history of AATD, we know that index cases, those seen by medical care facilities, are more ill compared with nonindex cases (family members) [26]. In a population-based cohort like CanCOLD, fewer individuals have developed pulmonary symptoms and associated inflammatory milieu that would favour disease progression in the intermediate AAT deficiency state. Finally, lower smoking exposure may also have limited expressivity.
SERPINA1 genotyping grouping used in this study cannot be directly compared with previous studies. To make a fair comparison, we have performed MZ-only analysis and showed lower diffusing capacity in MZ heterozygotes. This result seems consistent with previous studies that have showed that MZ individuals are characterised by a lower FEV1/FVC ratio and more radiographic emphysema [24], accelerated forced expiratory force at 25–75% of FVC (FEF25–75%) decline in the presence of smoking or obesity [25], lower lung function and CT-based emphysema [22], and lower lung function and greater COPD risk, especially in ever-smokers [23].
This study demonstrates that DNA sequencing is feasible in a large cohort. DNA sequencing provides a complete assessment of the gene causing AATD, namely SERPINA1. It can detect conventional protease inhibitor-deficient alleles (e.g. Z and S), but also rare and novel genetic variants. Diagnosis at the DNA level is the modern gold standard information needed for hereditary disorders. Importantly, DNA sequencing can be implemented in any laboratory with a standard DNA sequencing system, has a fast turnaround time, and may eventually replace the historical multistep testing algorithms to detect AATD used in most countries [9–11]. With established consistency in diagnosing AATD at the DNA level, there is potential to expedite the clinical decision making process recommended by many national and international lung societies for targeted testing and augmentation therapy [1–4].
In this study, we have not performed cost-effectiveness analysis of SERPINA1 DNA sequencing relative to current multistep-testing algorithms to detect AATD. For practical considerations, we provide costs for DNA sequencing. In Canadian currency, the cost of materials and supplies to sequence the 1359 CanCOLD participants is estimated at CAD 31 500 ((CAD 1.85 per PCR reaction+CAD 3.75 per sequencing reaction)×4 amplicons per sample×1359 samples). About CAD 10 per sample must be added for DNA extraction. An effective workforce and the number of samples processed at the same time are key to keep the cost low. It takes about a day and a half to complete the protocol. The salary of a laboratory technician for that time is thus the main factor fixing the cost per sample. In this research project, we were able to process samples in 96-well plate format and save costs. Lower throughput is expected in a realistic clinical healthcare system. For example, in our laboratory, the overall cost of SERPINA1 DNA sequencing per sample is CAD 650, which drops to CAD 150 when eight samples are processed at the same time. This implies that all equipment are in place, including a sequencer and about CAD 20 000 of ancillary equipment (thermocycler, centrifuges, pipettes, water bath, vortex and electrophoresis system). DNA sequencers come in different throughput formats and prices, but one that can process four samples at the time is available at approximately CAD 50 000. Sequencing reactions can also be outsourced if a sequencer is not available. All these costs must be balanced with the resulting benefits. Here, we tested 1359 individuals and obtained a conclusive AATD diagnostic for all of them, i.e. success rate=100%. The single step test can be performed in 2 days (more realistically offered within a week) and thus has the potential to end the diagnostic odyssey of patients with AATD.
This study has many strengths, including being the first Canadian COPD cohort that has recruited its participants from the general population rather than convenient sampling often utilised in clinical research. Furthermore, the research participants have been extensively phenotyped for COPD (clinical, physiology, CT scan with qualitative and quantitative evaluation). The limitations include the relatively limited sample size. As with other cohort studies, participation bias may affect the representativeness of the cohort relative to the underlying population. The current study has been limited to cross-sectional data. Longitudinal data collection is ongoing, and future analyses are needed with respect to COPD development and progression. We attempted to classify all 34 genetic variants identified in CanCOLD participants as normal or deficient (table 1). This was based on all possible sources of information including previous literature, location of the variants in the protein structure, pathogenicity scores and AAT serum levels among carriers. However, some uncertainties remain about the classification of rare variants and further functional studies will be needed. Finally, the four-group classification scheme used in this study (no, mild, intermediate and severe deficiency) that reflects the combination of the two alleles observed in each individual is likely to be challenged and refined with time. However, grouping genotypes has always been inherent (consciously or not) in the field of AATD. For example, how many individuals would be MM in CanCOLD based on the conventional protease inhibitor system? With DNA sequencing, we demonstrated that the no deficiency group (individuals with two normal alleles) is in fact a collection of 49 different genotypes (see abscissa of figure 4). Groups were thus formed in this study using an unprecedented level of granularity. The strong association and progressive decline in AAT levels with SERPINA1 genotyping groups (figure 5a) support our grouping scheme.
In conclusion, this study identified 34 genetic variants in the SERPINA1 gene resulting in 220 deficient alleles from 1359 individuals. Four AATD cases were identified and the remaining deficient alleles were carried by 206 individuals, indicating that deficient alleles affected 15.5% of subjects in this Canadian population-based cohort. Patients with genotypes resulting in severe AATD (AATD cases) are more susceptible to develop airway obstruction as demonstrated by lower diffusion capacity and greater CT-based emphysema. In contrast, mild and intermediate deficiency genotypes were not associated with COPD phenotypes in this random sampling cohort except a lower diffusion capacity in MZ heterozygotes. Overall, this study demonstrates the frequency of deficient alleles beyond the most common S and Z alleles in a population-based cohort, and the feasibility of DNA sequencing on a large scale to provide an accurate and definitive diagnosis for AATD. The longitudinal evaluation will be important to determine the effects of SERPINA1 genotypes on COPD risk and progression.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00958-2020.SUPPLEMENT
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00958-2020.Shareable
Acknowledgements
The authors thank the men and women who participated in the study and individuals in the CanCOLD Collaborative research Group. Executive Committee: Jean Bourbeau (McGill University, Montreal, QC, Canada); Wan C. Tan, J. Mark FitzGerald, Don D. Sin (UBC, Vancouver, BC, Canada); Darcy D. Marciniuk (University of Saskatoon, Saskatoon, SK, Canada); Denis E. O'Donnell (Queen's University, Kingston, ON, Canada); Paul Hernandez (University of Halifax, Halifax, NS, Canada); Kenneth R. Chapman (University of Toronto, Toronto, ON, Canada); Robert Cowie (University of Calgary, Calgary, AB, Canada); Shawn Aaron (University of Ottawa, Ottawa, ON, Canada); François Maltais (University of Laval, Quebec City, QC, Canada). International Advisory Board: Jonathon Samet (the Keck School of Medicine of USC, California, USA); Milo Puhan (John Hopkins School of Public Health, Baltimore, USA); Qutayba Hamid (McGill University, Montreal, QC, Canada); James C. Hogg (University of British Columbia, James Hogg Research Center, Vancouver, BC, Canada). Operations Center: Jean Bourbeau (PI), Carole Baglole, Carole Jabet, Palmina Mancino, Yvan Fortier (University of McGill, Montreal, QC, Canada); Wan C. Tan (co-PI), Don Sin, Sheena Tam, Jeremy Road, Joe Comeau, Adrian Png, Harvey Coxson, Miranda Kirby, Jonathon Leipsic, Cameron Hague (University of British Columbia, James Hogg Research Center, Vancouver, BC, Canada). Economic Core: Mohsen Sadatsafavi (University of British Columbia, Vancouver, BC). Public Health Core: Teresa To, Andrea Gershon (University of Toronto). Data management and Quality Control: Wan C. Tan, Harvey Coxson (UBC, Vancouver, BC, Canada); Jean Bourbeau, Pei-Zhi Li, Jean-Francois Duquette, Yvan Fortier, Andrea Benedetti, Denis Jensen (McGill University, Montreal, QC, Canada); Denis O'Donnell (Queen's University, Kingston, ON, Canada). Field Centers: Wan C. Tan (PI), Christine Lo, Sarah Cheng, Elena Un, Cindy Fung, Wen Tiang Wang, Faize Faroon, Olga Radivojevic, Carl Zou (UBC James Hogg Research Center, Vancouver, BC, Canada); Jean Bourbeau (PI), Palmina Mancino, David Latreille, Jacinthe Baril, Laura Labonte (McGill University, Montreal, QC, Canada); Kenneth Chapman (PI), Patricia McClean, Nadeen Audisho (University of Toronto, Toronto, ON, Canada); Brandie Walker, Robert Cowie (PI), Ann Cowie, Curtis Dumonceaux, Lisette Machado (University of Calgary, Calgary, AB, Canada); Paul Hernandez (PI), Scott Fulton, Kristen Osterling (University of Halifax, Halifax, NS, Canada); Shawn Aaron (PI), Kathy Vandemheen, Gay Pratt, Amanda Bergeron (University of Ottawa, Ottawa, ON, Canada); Denis O'Donnell (PI), Matthew McNeil, Kate Whelan (Queen's University, Kingston, ON, Canada); François Maltais (PI), Cynthia Brouillard (University of Laval, Quebec City, QC, Canada); Darcy Marciniuk (PI), Ron Clemens, Janet Baran (University of Saskatoon, Saskatoon, SK, Canada).
Footnotes
This article has an editorial commentary: https://doi.org/10.1183/13993003.02628-2020
This article has supplementary material available from erj.ersjournals.com
Author contributions: Y. Bossé and J. Bourbeau conceived the study, have full access to the data, and take responsibility for the integrity of the data and article. N. Gaudreault and C. Henry performed the DNA sequencing. S. Thériault measured the AAT serum levels. M. Kirby, F. Maltais W. Tan and J. Bourbeau contributed to the study design, performed clinical recruitment and phenotyping of patients. P.Z. Li and Y. Bossé performed data analysis. N. Gupta, J. Bourbeau and Y. Bossé wrote the manuscript. N. Gaudreault, S. Thériault, M. Kirby, F. Maltais and W. Tan edited the manuscript for intellectual content. All authors approved the final manuscript.
Conflict of interest: N. Gupta has nothing to disclose.
Conflict of interest: N. Gaudreault has nothing to disclose.
Conflict of interest: S. Thériault has nothing to disclose.
Conflict of interest: P.Z. Li has nothing to disclose.
Conflict of interest: C. Henry has nothing to disclose.
Conflict of interest: M. Kirby is a consultant for Vida Diagnostics Inc., outside the submitted work.
Conflict of interest: F. Maltais reports grants from AstraZeneca and GlaxoSmithKline, Boehringer Ingelheim, GSK, Sanofi and Novartis during the conduct of this study, and personal fees for serving on speaker bureaus and consultation panels from Boehringer Ingelheim, Grifols and Novartis, outside the submitted work; and is financially involved with Oxynov, a company which is developing an oxygen delivery system.
Conflict of interest: W. Tan reports grants from Canadian Institute of Heath Research (CIHR/Rx&D Collaborative Research Program Operating Grants- 93326) with industry partners AstraZeneca Canada Ltd, Boehringer Ingelheim Canada Ltd, GlaxoSmithKline Canada Ltd, Merck, Novartis Pharma Canada Inc., Nycomed Canada Inc. and Pfizer Canada Ltd, during the conduct of the study.
Conflict of interest: J. Bourbeau reports grants from CIHR, Canadian Respiratory Research Network (CRRN), Foundation of the MUHC and Aerocrine, personal fees for consultancy and lectures from Canadian Thoracic Society and CHEST, grants and personal fees for advisory board work and lectures from AstraZeneca, Boehringer Ingelheim, Grifols, GlaxoSmithKline, Novartis and Trudell, outside the submitted work.
Conflict of interest: Y. Bossé reports grants from Grifols Canada Ltd, during the conduct of the study; personal fees for lectures from Grifols Canada Ltd, outside the submitted work.
Support statement: This work was supported by Grifols Canada Ltd, the Fondation de l'Institut universitaire de cardiologie et de pneumologie de Québec, the Respiratory Health Network of the Fonds de recherche Québec – Santé (FRQS), and the Canadian Institutes of Health Research (MOP - 123369). The Canadian Cohort Obstructive Lung Disease (CanCOLD) study is currently funded by the Canadian Respiratory Research Network (CRRN); industry partners: AstraZeneca Canada Ltd; Boehringer Ingelheim Canada Ltd; GlaxoSmithKline Canada Ltd; Novartis. Researchers at RI-MUHC Montreal and Icapture Centre Vancouver lead the project. Previous funding partners are the CIHR (CIHR/Rx&D Collaborative Research Program Operating Grants- 93326); the RHN of the FRQS; industry partners: Almirall; Merck Nycomed; Pfizer Canada Ltd; and Theratechnologies. S. Thériault holds a Junior 1 Clinical Research Scholar award from the FRQS. M. Kirby holds a Canada Research Chair in Quantitative Imaging. F. Maltais holds a GSK Research Chair on COPD at Université Laval. J. Bourbeau holds a GSK/CIHR Research Chair on COPD at McGill University. Y. Bossé holds a Canada Research Chair in Genomics of Heart and Lung Diseases. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 1, 2020.
- Accepted May 20, 2020.
- Copyright ©ERS 2020
References










