





Abstract
Objective There is a paucity of observational data on antifibrotic therapy for idiopathic pulmonary fibrosis (IPF). We aimed to assess the course of disease of IPF patients with and without antifibrotic therapy under real-life conditions.
Methods We analysed data from a non-interventional, prospective cohort study of consecutively enrolled IPF patients from 20 interstitial lung disease expert centres in Germany. Data quality was ensured by automated plausibility checks, on-site monitoring, and source data verification. Propensity scores were applied to account for known differences in baseline characteristics between patients with and without antifibrotic therapy.
Results Among the 588 patients suitable for analysis, the mean±sd age was 69.8±9.1 years, and 81.0% were male. The mean±sd duration of disease since diagnosis was 1.8±3.4 years. The mean±sd value at baseline for forced vital capacity (FVC) and diffusion capacity (DLCO) were 68.6±18.8% predicted and 37.8±18.5% predicted, respectively. During a mean±sd follow-up of 1.2±0.7 years, 194 (33.0%) patients died. The 1-year and 2-year survival rates were 87% versus 46% and 62% versus 21%, respectively, for patients with versus without antifibrotic therapy. The risk of death was 37% lower in patients with antifibrotic therapy (hazard ratio 0.63, 95% CI 0.45; 0.87; p=0.005). The results were robust (and remained statistically significant) on multivariable analysis. Overall decline of FVC and DLCO was slow and did not differ significantly between patients with or without antifibrotic therapy.
Conclusions Survival was significantly higher in IPF patients with antifibrotic therapy, but the course of lung function parameters was similar in patients with and without antifibrotic therapy. This suggests that in clinical practice, premature mortality of IPF patients eventually occurs despite stable measurements for FVC and DLCO.
Abstract
Survival was significantly higher in antifibrotic-treated (AT) IPF patients, but the course of lung function parameters was similar in AT and non-AT patients, suggesting that functional stability alone may not safeguard against premature mortality in IPF https://bit.ly/2RDsrVY
Introduction
Idiopathic pulmonary fibrosis (IPF) is a severe respiratory disease characterised by progressive scarring of the lung, leading to respiratory failure and death within 3–5 years from diagnosis [1]. Effective treatments are still limited. The antifibrotic treatments pirfenidone and nintedanib have been shown to slow disease progression as measured by annual rate of decline in forced vital capacity (FVC) [2], but their effect on lung function and survival under clinical practice conditions warrants further exploration.
As randomised controlled studies on antifibrotic treatments have limitations in terms of their generalisability due to patient selection/exclusion and duration of follow-up, observational data in unselected IPF patients are needed to provide a more comprehensive picture. A number of registries have been initiated in various countries to provide such real-life data [3–8], but their follow-up is limited to 1–2 years only.
The database of the INSIGHTS-IPF (Investigating Significant Health Trends in Idiopathic Pulmonary Fibrosis) registry, one of the largest IPF registries worldwide, offers the opportunity to analyse the course of disease and long-term effectiveness of antifibrotic therapy in IPF. The aims of the present analysis were 1) to describe and compare cohorts of patients with and without antifibrotic therapy; 2) to assess the correlation between antifibrotic drug use and lung function; and 3) to test the correlation between antifibrotic drug use and survival.
Methods
Design and parameters
The INSIGHTS-IPF registry is a nationwide, investigator-initiated observational study. The registry has been continuously enrolling consecutive incident and prevalent patients in routine clinical care across 20 pulmonary specialist centres in Germany since November 2012. Patients aged ≥18 years with a study-site diagnosis of IPF according to the 2011 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association IPF guideline [9] after provision of written informed consent can be enrolled, with no explicit exclusion criteria. The registry's structure, methodology, and regulatory aspects, as well as a detailed description of the baseline characteristics of the patient cohort, have been reported previously [10–12]. The study has been approved by the ethics committee at the Technical University of Dresden and various local ethical committees. All patients provided informed consent before their data were documented in the registry.
Data were collected at enrolment (baseline) and at subsequent 6- to 12-month intervals. At each follow-up visit, all clinical events, including hospitalisation and acute exacerbations (as judged by the treating physician), as well as deaths that occurred during the study period, were recorded by each site. At each visit, if available, a range of routine pulmonary function tests were documented, including FVC, diffusing capacity of the lung for carbon monoxide (DLCO), the forced expiratory volume in 1 s (FEV1) and 6-min walk distance (6MWD). The gender, age and physiology (GAP) index was calculated based on available data [13].
The treating physician was requested to judge the overall clinical course of IPF at baseline and each follow-up visit by the categories stable disease, slow progression, rapid progression, no judgement possible. Physiological changes between baseline and 2-year follow-up were categorised as stable if FVC did not change or was improved by ≥5%; as a moderate decrease if decreased by >5–10%; or as a significant decrease if decreased by >10%.
Quality measures
All data were collected using a standardised internet-based case report form with secure electronic data transfer to the central database. Quality measures included automated plausibility checks at data entry, statistical checks on data quality (focusing on missing values and outliers) as well as on-site monitoring and source data verification performed in the majority of centres (>70%).
Data analysis
Data were summarised by descriptive statistics including mean±sd and absolute and relative frequencies at baseline and each subsequent follow-up assessment. Data analysis comprised the period between the first documentation in the registry in December 2012 until the data cut-off point in December 2018. The analyses follow the intention-to-treat principle, which means that each patient with at least one dose of antifibrotic therapy is assigned to the treatment group.
The entire observation period was considered for each patient in the registry in order to compare outcomes, in terms of mortality and pulmonary function test results, between patients who were treated with antifibrotic therapy and those who were not. Patients in the registry who had never been treated with an antifibrotic therapy were assigned to the control group. The first observation in that group was the registry enrolment visit. Patients who started an antifibrotic therapy before enrolment into the registry (start date >10 days before, e.g. as participant in a clinical study) were excluded because of the nonavailability of clinical data at treatment start. The data were divided into individual treatment episodes for patients who started pirfenidone and/or nintedanib during the observation period. For these patients, the first observation was the initial treatment visit. If a patient was treated with pirfenidone and nintedanib (in sequence) during follow-up, then two treatment episodes were assigned (one for each drug) for the pulmonary function and 6MWD tests at the corresponding time point. In contrast, the risk of mortality was analysed for the last available antifibrotic treatment episode in patients who were treated with pirfenidone followed by nintedanib, or vice versa, during follow-up. All patients with a follow-up period of ≥3 months were included in the analyses. In addition, a follow-up interval of 2 years was considered. The primary analysis for lung function tests and 6MWD is based on the observed values in the registry. Since the number of missing values in lung function tests (FVC baseline 4.5%, follow-up 20.7%; DLCO baseline 16.2%, follow-up 31.9%) and 6MWD (baseline 14.1%, follow-up 57.3%) were substantial, we applied the technique of multiple imputation for those variables to estimate the missing values as sensitivity analyses. Patients with a missing lung function test tended to be on a less severe disease course compared to patients with available lung function test. Preliminary analyses showed that mortality, age and comorbidities were associated with the absence of the considered variables. Therefore, the first sensitivity analysis used an imputation model including the predictor variables age, sex, number of comorbidities, IPF duration, mortality, antifibrotic therapy and the lung function and 6MWD results from the prior visit. The number of imputations was set to 10. As a second sensitivity analysis, the last observation carried forward method for lung function and 6MWD was used as well. The third sensitivity analysis used the imputation of missing values by the worst possible value (FVC, DLCO and 6MWD of 0) for patients who died.
Propensity score
INSIGHTS-IPF is an observational study and thus allocation to treatment was not randomly assigned. Consequently, various patient characteristics at baseline may be imbalanced, possibly leading to biased results and conclusions. The standard approach to deal with this problem is to model the probability of treatment assignment by the physician (propensity score) based on the clinical characteristics at treatment start in order to balance the characteristics of the two considered groups of patients [14–16]. The propensity score was estimated by a logistic regression model that included the covariates sex, age, smoking status, number of comorbid diseases, IPF disease duration, FVC % predicted, 6MWD, concomitant therapy with steroids and the global assessment of the disease course by the physician at baseline. A weight value (inverse probability of treatment weighting) was calculated for each patient based on the propensity score [17]. All statistical comparisons between patients with and without antifibrotic therapy were weighted to balance the two groups regarding the clinical characteristics at treatment start.
In the primary analysis, the course of the pulmonary function (FVC % pred and DLCO % pred) and 6MWD tests were analysed by weighted linear mixed models to account for the possibility of two treatment episodes for a single patient (additional cluster variable) and the longitudinal study design based on the observed values. An interaction term treatment × time was included into the weighted linear mixed models to test for differences in change in the three considered parameters by treatment. Secondary analyses of lung function and 6MWD included the imputed data, which employed two imputation methods: last observation carried forward and worst-case imputation. The risk of mortality was analysed by a multivariable Cox proportional hazard model weighted by the propensity score. The proportional-hazards assumption was tested on the basis of Schoenfeld residuals after fitting the Cox regression model.
Data were analysed using Stata 12.1 (Stata Statistical Software: release 12; StataCorp LP, College Station, TX, USA).
Results
588 patients were deemed suitable for the present analysis. The mean age of the study population was 69.8 years, with a large male preponderance (81.0%). The mean±sd duration of symptoms before the baseline visit was 3.5±4.2 years and time between diagnosis and study enrolment was 1.8±3.4 years. 58% of the patients had disease duration of <12 months and 47% of <6 months. The mean Borg dyspnoea score was 2.2±2.4, and the GAP index stages were as follows: stage I in 20.4% of the patients, stage II in 49.9% of the patients and stage III in 29.7% of the patients. In terms of lung function parameters at baseline, the mean±sd FVC was 68.6±18.8% pred and the DLCO was 37.8±18.5% pred. Health-related quality of life as measured on the 100-point visual analogue scale was 59.6±23.6. As current therapy at baseline, prednisone was reported in 23.6% and N-acetylcysteine in 25.5% of patients.
The mean±sd follow-up time was 1.2±0.7 years (maximum of 2 years) for the total sample, 1.2±0.5 years for patients receiving antifibrotic therapy and 1.0±0.7 years for patients who had never been treated with antifibrotic therapy. 334 treatment episodes under antifibrotic therapy (168 pirfenidone, 166 nintedanib) were reported for 298 patients in our registry, resulting in 36 (12%) patients with two episodes. Among these, pirfenidone was the first antifibrotic drug in 29 patients. Seven patients switched from pirfenidone to nintedanib within 3 months after discontinuation of pirfenidone; the other 22 patients started nintedanib on average 13 months after discontinuation of pirfenidone (table 1).
Characteristics of patients in the total analysed cohort and by presence or absence of antifibrotic treatment
Generalised linear mixed models were used to analyse the pulmonary function and 6MWD tests. These models included all antifibrotic therapy treatment episodes, and were based on the observed values. During the 2 years of follow-up, mean FVC % pred remained almost stable (figure 1ai; β for change in follow-up −0.42, 95% CI −1.44–0.60; p=0.416), with no significant differences between the two groups (β for time × therapy −0.65, 95% CI −1.82–0.52; p=0.274). Predicted DLCO showed a similar course in both groups (figure 1bi), with no significant decline in DLCO (β for change in follow-up −1.05, 95% CI −2.40–0.30; p=0.127) in follow-up and no significant differences between the two groups (β for time × therapy −0.40, 95% CI −2.56–1.77; p=0.721). Results for the 6MWD test were available in 89% of patients at baseline; however, this measurement was compromised by a high rate of missing data during follow-up. There was no statistically significant difference in the course of 6MWD results over time (β for change in follow-up −14.8, 95% CI −25.6–4.1; p=0.076), considering the observed values (figure 1ci). The primary analysis was repeated in patients with disease duration of ≤12 months at enrolment (prevalent patients, figure 1a-c,ii). A slightly better course of FVC % pred, DLCO % pred and 6MWD was observed in patients with antifibrotic therapy; however, the difference was not statistically significant. The sensitivity analyses using imputed data and data obtained by the last observation carried forward approach resulted in comparable results to those of the primary analysis. If an FVC of 0% was imputed in patients who died during follow-up, patients never on antifibrotic therapy tended to have a slightly, but not significantly stronger FVC decline. The decline in DLCO was worse in patients with antifibrotic treatment, although when imputation of the worst individual value was implemented, there were no significant differences between groups.
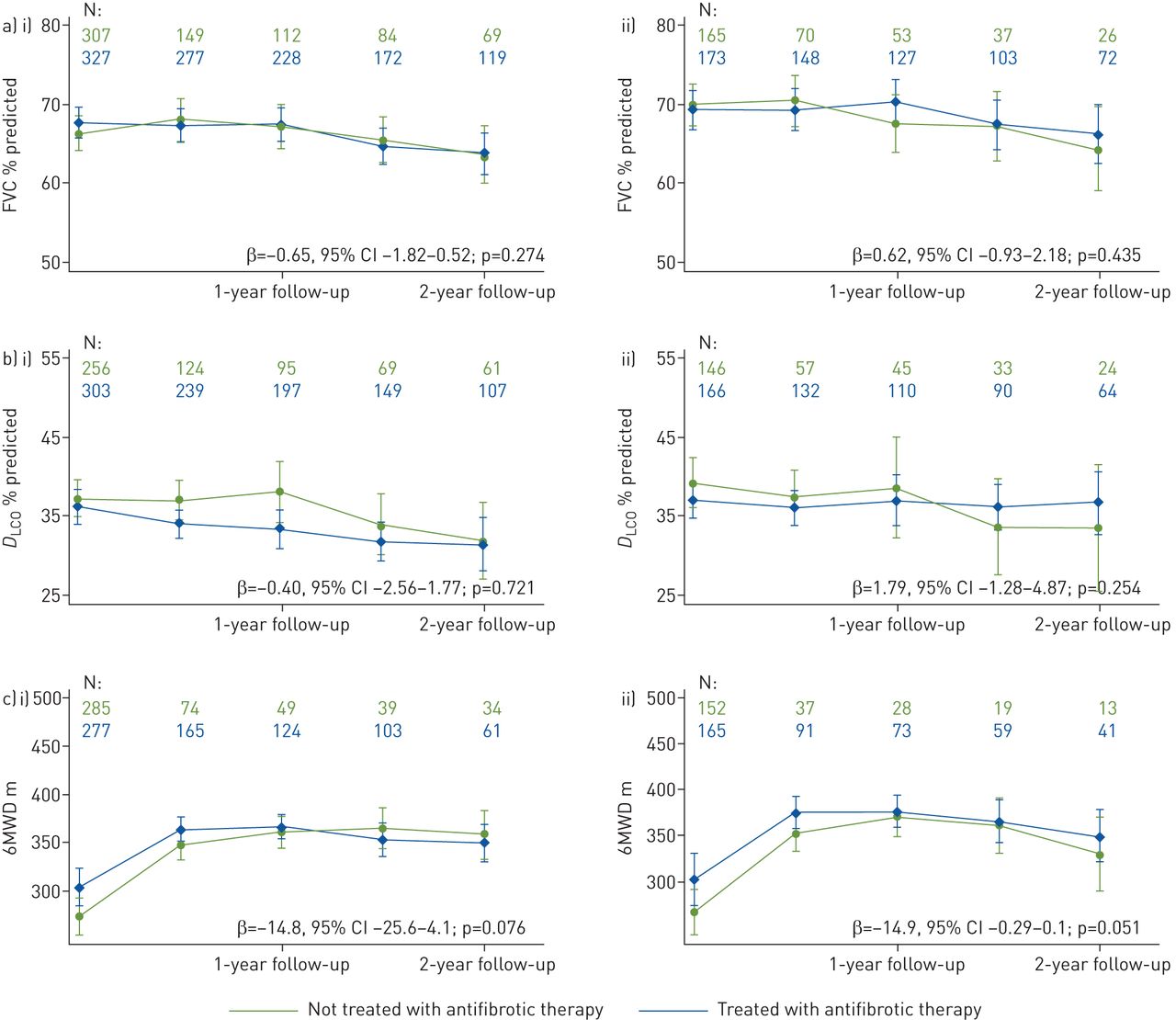
Change in a) forced vital capacity (FVC) % predicted, b) diffusing capacity of the lung for carbon monoxide (DLCO) % predicted and c) 6-min walk distance (6MWD) over the 2-year follow-up (β=interaction term time × therapy to estimate the difference in change during 2-year follow-up in the considered parameter between patients with and without antifibrotic therapy). i) All patients; ii) patients with a disease duration <12 months at enrolment.
The risk of mortality was analysed for the last available treatment episode in patients who were treated with pirfenidone (n=139) and nintedanib (n=159) in follow-up. A total of 194 (33.0%) patients died during follow-up. A total of 79 (41%) patients died of IPF-related reasons (20% respiratory failure, 8% respiratory infection/pneumonia), followed by complicating comorbidity (8%) and other causes not related to IPF (9%). The reason of death was unknown for 71 (37%) patients.
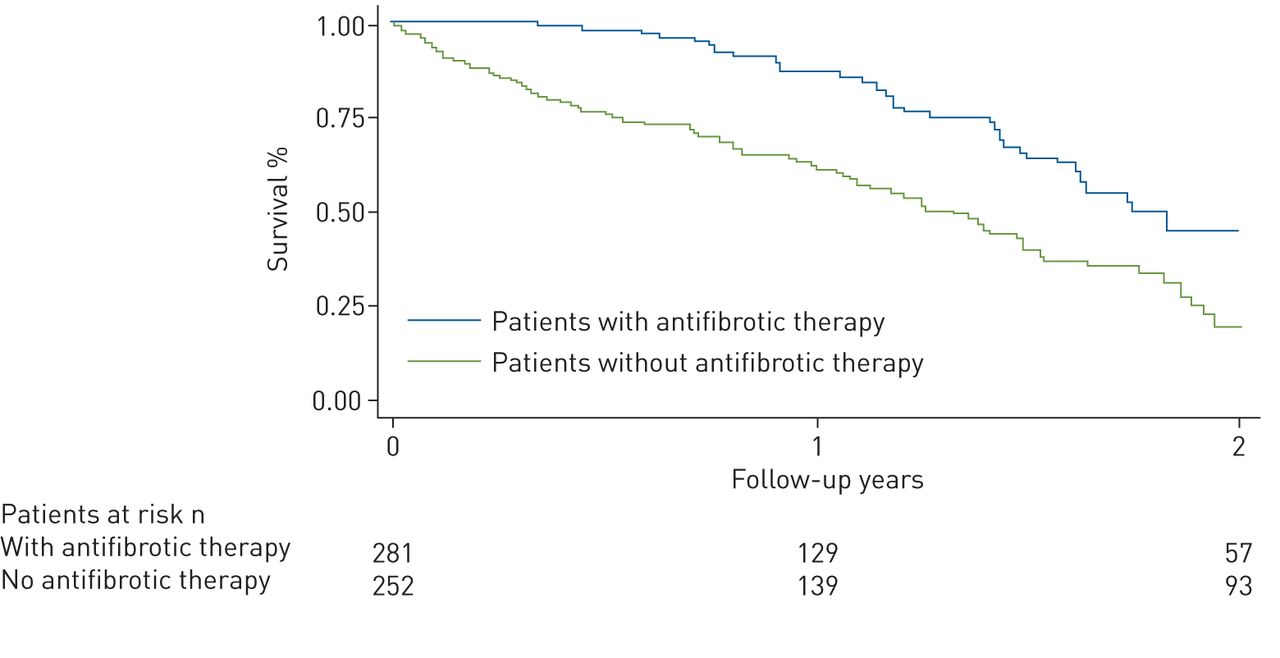
Overall mortality was substantially lower in patients treated with antifibrotic therapy. The risk of death for any reason was 37% lower in patients with antifibrotic therapy compared with those without such therapy (hazard ratio (HR) 0.63, 95% CI 0.45–0.87; p=0.005; figure 2). This result was robust (and remained statistically significant) on multivariable analysis, as reported in table 2. Analysis for both antifibrotic drugs approved for treating IPF, nintedanib and pirfenidone, revealed no statistically significant difference in overall mortality between the two drugs (HR for pirfenidone versus nintedanib 1.39, 95% CI 0.87–2.22; p=0.164).
{kind=link}
{kind=link}
Risk of mortality within 2 years by antifibrotic treatment (by propensity score weighted Kaplan–Meier survival curves).
Risk of mortality estimated by a multivariable Cox regression model
In patients treated with antifibrotics, the risk of IPF-related death was not (statistically significantly) lower compared to patients without such therapy (HR 0.75, 95% CI 0.45–1.25; p=0.266), while the risk of death for unknown reason was 56% lower in patients with antifibrotics (HR 0.44, 95% CI 0.26–0.75; p=0.003). Due to the lower numbers of events in this subgroup analysis, this result should be interpreted with caution.
We tested the hypothesis whether survival differs between patients with stable FVC (i.e. ≤10% decline during follow-up) compared to patients with worsening of FVC of >10% during follow-up, regardless of therapy. The risk of mortality was slightly higher in such patients with disease progression compared to stable IPF patients (HR 1.34, 95% CI 0.89–2.02; p=0.163). This result was confirmed while adjusting for the effect for antifibrotic treatment.
The risk of mortality was additionally analysed in patients with disease duration of <12 months prior to study enrolment. The risk of death in the subsample of incident patients was 64% lower in patients treated with antifibrotic therapy compared to controls (HR 0.44, 95% CI 0.25–0.78; p=0.003). The result was confirmed in multivariable analysis.
Discussion
The present analysis of the large and contemporary INSIGHTS-IPF registry indicates that patients on antifibrotic therapy appear to survive significantly longer than IPF patients without antifibrotic therapy. The lower overall mortality risk in the patients treated with antifibrotic medication was mainly driven by patients with unknown cause of death. The statistically nonsignificant relationship between antifibrotic therapy and IPF-related deaths might be due to the low number of recorded IPF-related deaths (79.4% of deaths).
Compared with the recently published observational data from the EurIPF registry, patients in INSIGHTS-IPF were nearly identical in terms of total lung capacity (70.0% pred versus 71.2% pred), FVC (68.4% pred versus 68.3% pred) and FEV1 (110% pred versus 111% pred), while DLCO was lower in our study (42.1% pred versus 37.8% pred) [18]. A subset of IPF patients with long-term follow-up within the EurIPF registry were analysed by Kaplan–Meier analysis (without propensity score matching) in correlation with the date of first IPF diagnosis. The analysis of this subset found that median survival on antifibrotic drugs was 123.1 months (censored cases inclusive, range 84–162 months), compared with a median survival of 68.3 months in patients treated with any other medication including immunosuppressive therapies (censored cases inclusive, range 54–83 months). Functional follow-up data from the EurIPF registry were not reported. Another difference between our data and those of the EurIPF registry, besides the larger number of patients and the statistics applied in our cohort, is the fact that pirfenidone was used in the vast majority (83%) of the EurIPF registry cohort, while in our study population nintedanib and pirfenidone where almost equally distributed, slightly favouring nintedanib (53.3%).
Interestingly, we observed a similar stable course of lung function parameters (FVC and DLCO) over time in both groups, with and without antifibrotic therapy, while overall mortality was considerably higher in the group not treated with antifibrotics. At first glance, our data could provide a basis for a hypothesis that stable physiological measurements such as FVC and DLCO alone may not provide a safeguard against premature mortality in IPF. Lung function measurement every 6–12 months is common practice and thus employed in our registry. However, such measurements may be less sensitive to detect differences in the course of IPF compared to highly standardised serial measurements at shorter intervals, which are commonly applied in clinical trials. Moreover, missing lung function data may have contributed to blunt differences of the slope of FVC and DLCO decline between patients with and without antifibrotic therapy. In this context, it is noteworthy that hospital-based FVC measurements, compared with unsupervised daily home measurements, have been suggested to be less sensitive in detecting progression of fibrosis and in predicting subsequent prognosis [19]. However, a recent clinical treatment trial using daily home spirometry for the primary end-point revealed potential technical and practical obstacles associated with this methodology [20].
The phenomenon of emphysema blunting the decline of FVC in both groups may have contributed to this observation, but the prevalence of emphysema as reported by the investigators was low in both groups. In addition, the higher preponderance of steroid-treated patients in the group not treated with antifibrotics may be considered to potentially contribute to a higher mortality in this group. However, the mean prednisone dosage in our study (given to a quarter of patients in our study) was 14 mg·day−1. In the INPULSIS study the maximum dose was 15 mg·day−1 and in the ASCEND study, prednisone was only allowed if given for another indication [21, 22]. Nonetheless, we cannot exclude that unbalanced steroid medication has contributed to the observed difference. Finally, antioxidant drugs (N-acetylcysteine) were less commonly used in the antifibrotic therapy arm. The impact of these drugs on prognosis is still under discussion, and thus a bias cannot be fully excluded [23, 24]. In consideration of all the limitations our data should be taken as a signal of caution that stability of FVC and DLCO may not always protect from premature mortality in the absence of antifibrotic therapy in a fatal disease such as IPF. The common practice, still widely used, of withholding antifibrotic therapy from physiologically stable IPF patients may therefore set these patients on a path of increased risk of dying [25].
Another important aspect of our study is the fact that all patients were enrolled solely based on investigator judgement. Therefore, the cohort of patients enrolled included all the imponderabilities of diagnosis in this complex disease that occur in daily practice. The observed difference in survival in favour of antifibrotic therapy is an important argument for the clinical application of these drugs, even though a causative argument cannot be made from our study. This observation is therefore in accordance with recent clinical trials showing that antifibrotic therapies are effective in progressive fibrotic interstitial lung diseases other than IPF [20, 26, 27].
Our data do not identify a cause for the difference in overall mortality between patients with and without antifibrotic therapy. However, we can speculate that acute exacerbation may have contributed substantially to this difference.
A number of limitations need to be taken into consideration when interpreting the findings. The major limitation of this study is that patients with existing (prevalent) and newly diagnosed (incident) IPF were documented, which may potentially cause lead time bias regarding mortality. This is especially important since time to diagnosis was ∼1 year longer in the never-treated population, which could indicate a “healthy survivor effect” [28]. Furthermore, there was no randomisation between the group of patients who had never been treated with an antifibrotic therapy and patients who were treated with an antifibrotic drug. To account for bias by indication, we calculated a propensity score to estimate the probability of being treated with an antifibrotic drug in our registry based on clinical characteristics. However, there may exist unmeasured variables that cannot be included in the propensity score model that may have impacted the association between antifibrotic therapy and mortality. Furthermore, accompanying therapies such as anti-oxidant or anti-acid therapy may have impacted the results of our analysis. In addition, we had to account for a high proportion of missing values in the pulmonary function tests and in the 6MWD test in the follow-up data, which could have affected our results. The fact that only ILD specialty centres participated in the INSIGHTS-IPF registry may limit the generalisability of our study.
In conclusion, we were able to demonstrate a significant lower all-cause mortality in IPF patients treated with antifibrotic drugs when compared to a matched cohort of IPF patients not treated with antifibrotic drugs. Moreover, our analysis provides a basis for the hypothesis that stability of lung function parameters over time, especially FVC and DLCO, in untreated IPF patients may be misleading as our data indicate that stability of these parameters probably do not protect from premature death.
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02279-2019.Shareable
Acknowledgements
The authors thank the patients for their participation in the registry. The authors acknowledge the sustained valuable contribution of Silke Geier (deceased in September 2019) to this registry.
Footnotes
This study is registered at www.clinicaltrials.gov with identifier number NCT01695408. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Conflict of interest: J. Behr reports grants and personal fees from Boehriger Ingelheim, personal fees from Actelion, Roche, Galapagos, Promedior, BMS and MSD, during the conduct of the study.
Conflict of interest: A. Prasse reports grants and personal fees from Roche/InterMune and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: H. Wirtz reports personal fees from Roche and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: D. Koschel has nothing to disclose.
Conflict of interest: D. Pittrow reports personal fees from Actelion, Bayer, Boehringer Ingelheim, Sanofi, Biogen, Shield and MSD, outside the submitted work.
Conflict of interest: M. Held has nothing to disclose.
Conflict of interest: J. Klotsche has nothing to disclose.
Conflict of interest: S. Andreas reports grants and personal fees from Boehringer Ingelheim, personal fees from Roche, outside the submitted work.
Conflict of interest: M. Claussen reports personal fees from Roche and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: C. Grohé has nothing to disclose.
Conflict of interest: H. Wilkens reports personal fees from Boehringer Ingelheim, Roche, Actelion, Biotest, GSK, Pfizer and Bayer, outside the submitted work.
Conflict of interest: L. Hagmeyer has nothing to disclose.
Conflict of interest: D. Skowasch reports personal fees from Boehringer Ingelheim and Roche, outside the submitted work.
Conflict of interest: J.F. Meyer reports personal fees for lectures from Boehringer Ingelheim and Novartis, outside the submitted work.
Conflict of interest: J. Kirschner has nothing to disclose.
Conflict of interest: S. Gläser reports grants and personal fees from Boehringer Ingelheim and Novartis, personal fees from Roche, Actelion, Berlin Chemie and Astra, outside the submitted work.
Conflict of interest: N. Kahn has nothing to disclose.
Conflict of interest: T. Welte reports grants and personal fees from Boehringer Ingelheim, grants from Roche, outside the submitted work.
Conflict of interest: C. Neurohr reports personal fees from Boehringer Ingelheim and Roche, outside the submitted work.
Conflict of interest: M. Schwaiblmair has nothing to disclose.
Conflict of interest: T. Bahmer reports grants from German Center of Lung Research (DZL), personal fees from Roche, AstraZeneca, Chiesi, GSK and Novartis, outside the submitted work.
Conflict of interest: T. Oqueka has nothing to disclose.
Conflict of interest: M. Frankenberger has nothing to disclose.
Conflict of interest: M. Kreuter reports grants and personal fees from Roche/InterMune and Boehringer Ingelheim, outside the submitted work.
Support statement: This study and all costs associated with the development and publication of this manuscript were funded by Boehringer Ingelheim. The company has no influence on the conduct of the study or interpretation of data, or reporting. The study was supported with an unrestricted educational grant by Boehringer Ingelheim, Germany. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received November 26, 2019.
- Accepted April 7, 2020.
- Copyright ©ERS 2020
References










