





Abstract
Background In animal models of pulmonary arterial hypertension (PAH), angiotensin-converting enzyme (ACE)2 and angiotensin (Ang)-(1–7) have been shown to have vasodilatory, antiproliferative, antifibrotic and antihypertrophic properties. However, the status and role of the ACE2-Ang(1–7) axis in human PAH is incompletely understood.
Methods We studied 85 patients with a diagnosis of PAH of distinct aetiologies. 55 healthy blood donors paired for age and sex served as controls. Blood samples were obtained from the pulmonary artery in patients with PAH during right heart catheterisation. Peripheral blood was obtained for both groups. Ang(1–7) and -II were measured using zone capillary electrophoresis. Aldosterone, Ang(1–9), AngA and ACE2 were measured using ELISA, and ACE2 activity was determined enzymatically.
Results Of the 85 patients, 47 had idiopathic PAH, 25 had PAH associated with congenital heart disease and 13 had PAH associated with collagen vascular disease. Compared to controls, patients with PAH had a higher concentration of AngII (median 1.03, interquartile range 0.72–1.88 pmol·mL−1 versus 0.19, 0.10–0.37 pmol·mL−1; p<0.001) and of aldosterone (88.7, 58.7–132 ng·dL−1 versus 12.9, 9.55–19.9 ng·dL−1; p<0.001). Conversely, PAH patients had a lower concentration of Ang(1–7) than controls (0.69, 0.474–0.91 pmol·mL−1 versus 4.07, 2.82–6.73 pmol·mL−1; p<0.001), and a lower concentration of Ang(1–9) and AngA. Similarly, the ACE2 concentration was higher than in controls (8.7, 5.35–13.2 ng·mL−1 versus 4.53, 1.47–14.3 ng·mL−1; p=0.011), whereas the ACE2 activity was significantly reduced (1.88, 1.08–2.81 nmol·mL−1 versus 5.97, 3.1–17.8 nmol·mL−1; p<0.001). No significant differences were found among the three different aetiological forms of PAH.
Conclusions The AngII–ACE2–Ang(1–7) axis appears to be altered in human PAH and we propose that this imbalance, in favour of AngII, plays a role in the pathogenesis of the severe PAH. Further mechanistic studies are warranted.
Abstract
This study demonstrates that in patients with PAH of different aetiologies there are alterations of the ACE2-angiotensin (1–7)-MAS axis. Analysis of blood samples also demonstrates the presence of antibodies directed against ACE2 https://bit.ly/3alEbnJ
Introduction
Pulmonary arterial hypertension (PAH) encompasses a group of diseases characterised by a broad spectrum of pulmonary vascular changes leading to elevated pulmonary artery pressures, right heart failure (RHF) and untimely death [1,2]. Despite advances in its treatment, RHF and a low cardiac output state are almost inevitable outcomes in the natural history of PAH [3–5]. It has been demonstrated that the sympathetic nervous system as well as the renin–angiotensin–aldosterone system (RAAS) are hyperactivated in PAH and may negatively impact survival [4, 5]. A mechanistic role of sympathetic overdrive and hyperactive RAAS in the development of PAH has also been postulated [6–9].
The angiotensin-converting enzyme (ACE)2 converts angiotensin (Ang)II into Ang(1–7), perhaps to counterbalance the deleterious vascular effects of AngII [10–12]. Both in vitro studies and animal models of PAH have shown that Ang(1–7) has antiproliferative, antifibrotic and antihypertrophic properties, in addition to its vasodilatory effects [13, 14]. Based on this knowledge, recent attempts to favourably modify the ACE2–Ang(1–7) axis have been undertaken [15], despite incomplete knowledge of this system in human PAH. Here, we sought to further characterise the Ang II–ACE2–Ang(1–7) axis in PAH patients and hypothesised that, compared to healthy controls, patients with PAH will have higher levels of AngII, lower expression of ACE2 and therefore lower levels of Ang(1–7). In addition, we explored the serum concentration of other novel protective angiotensins, such as Ang(1–9) and AngA-alamandine in this population.
Material and methods
Study population
We studied 85 patients with PAH of diverse aetiologies (median, interquartile range (IQR) age 34, 25.5–48.0 years; 85% female) recruited from and followed by the pulmonary hypertension clinic of the cardiopulmonary department of the National Institute of Cardiology of Mexico. The diagnosis of PAH was established in accordance with current guidelines for diagnosis and treatment of PAH [1]. All patients underwent a thorough diagnostic workup including right heart catheterisation (RHC). The diagnosis of PAH was established by exclusion of secondary causes of pulmonary hypertension and demonstration of a mean pulmonary artery pressure (mPAP) ≥25 mmHg at rest, a pulmonary capillary wedge pressure (PCWP) ≤15 mmHg and a pulmonary vascular resistance >3 Wood units by RHC [1]. 55 carefully selected healthy blood-donors, free from cardiovascular disease, from our institutional blood transfusion biobank, paired for age and sex, were used as controls. During the diagnostic RHC of PAH patients a blood sample was obtained from the pulmonary artery and from a peripheral (cubital or vena cava) vein for enzyme and peptide measurements. Only peripheral blood was available for controls. Blood samples were processed immediately. The investigation and ethics committees of the Ignacio Chávez National Heart Institute (Mexico City, Mexico) approved the study, and each participant (patients and controls) gave an informed consent. Methods for the measurements of AngII and Ang(1–7) [16], Ang(1–9), and Angiotensin A levels, as well as ACE2 concentration and ACE2 activity are described in the supplementary material.
Statistical analysis
We compared Ang II, aldosterone, Ang(1–7), ACE2 levels, ACE2 activity, Ang(1–9) and AngA concentration measurements between PAH patients and controls. In addition, for patients, we compared baseline demographic characteristics, laboratory, echocardiography, haemodynamic and enzyme and peptide findings among the three aetiologic subgroups of PAH. Finally, we compared enzymatic measurements between pulmonary and peripheral vein samples within every single patient.
All data were verified for normal distribution by Shapiro–Wilks test. All categorical data were summarised as frequencies and percentages. Continuous variables were reported as medians and 25th and 75th percentiles (IQR). Statistical differences between groups were assessed, either using the Chi-squared or Fisher's exact test in the case of categorical variables. For continuous variables, we used the Kruskal–Wallis or Mann–Whitney U-tests, as appropriate. We performed the Kruskal–Wallis test with Dunn's nonparametric pairwise post hoc test with Bonferroni corrections to assess group differences. The Wilcoxon signed-rank test was used to compare paired samples. A p-value <0.05 was considered significant. Data were analysed using IBM SPSS Statistics for Windows (version 23.0; IBM Corp., Armonk, NY, USA).
Results
The clinical, laboratory, echocardiography and haemodynamic characteristics of the 85 studied PAH patients are summarised in table 1. All patients had severe PAH, as evidenced by mPAP of 56 (43.5–70) mmHg. All PAH patients had a normal PCWP, and the median (IQR) cardiac index was 2.76 (2.31–4.20) L·min−1·m−2. 37 (43.5%) of the 85 patients were in World Health Organization (WHO) functional class III and IV and had a baseline 6-min walk distance (6MWD) of 316 (IQR 242–389) m. 47 patients were diagnosed with idiopathic PAH (IPAH), 13 patients had PAH associated with collagen vascular disease (PAH-CTD) and 25 had PAH associated with congenital heart disease (PAH-CHD). Differences in demographics and clinical variables among subtypes of PAH are shown in table 1. Haemodynamically, all three groups were similar, except for a higher right atrial pressure (RAP) and PCWP in PAH-CHD patients, and the pulmonary artery pressures were lower in the PAH-CTD group. Prior to the baseline RHC, 52 (61%) patients had been treated with specific PAH drugs including sildenafil, endothelin-receptor antagonists (ERAs) and prostanoids, either as monotherapy (n=32) or as a combination therapy (n=20). 29 patients were treated with sildenafil alone, seven were receiving sildenafil plus an ERA, six were receiving sildenafil plus calcium channel-blockers, five were treated with sildenafil plus treprostinil, three were on calcium channel blockers (CCB) alone, and two patients were receiving sildenafil plus an ERA and a CCB. 24 (28.2%) patients were taking spironolactone, and 11 (12.9%) had received either ACE inhibitors or angiotensin-receptor antagonists. All these medications were discontinued between 8 and 24 h prior to the RHC.
Demographic, clinical, functional, laboratory, echocardiography and haemodynamic characteristics of pulmonary arterial hypertension (PAH) patients
Peptide concentrations and enzyme activity differences between PAH and controls
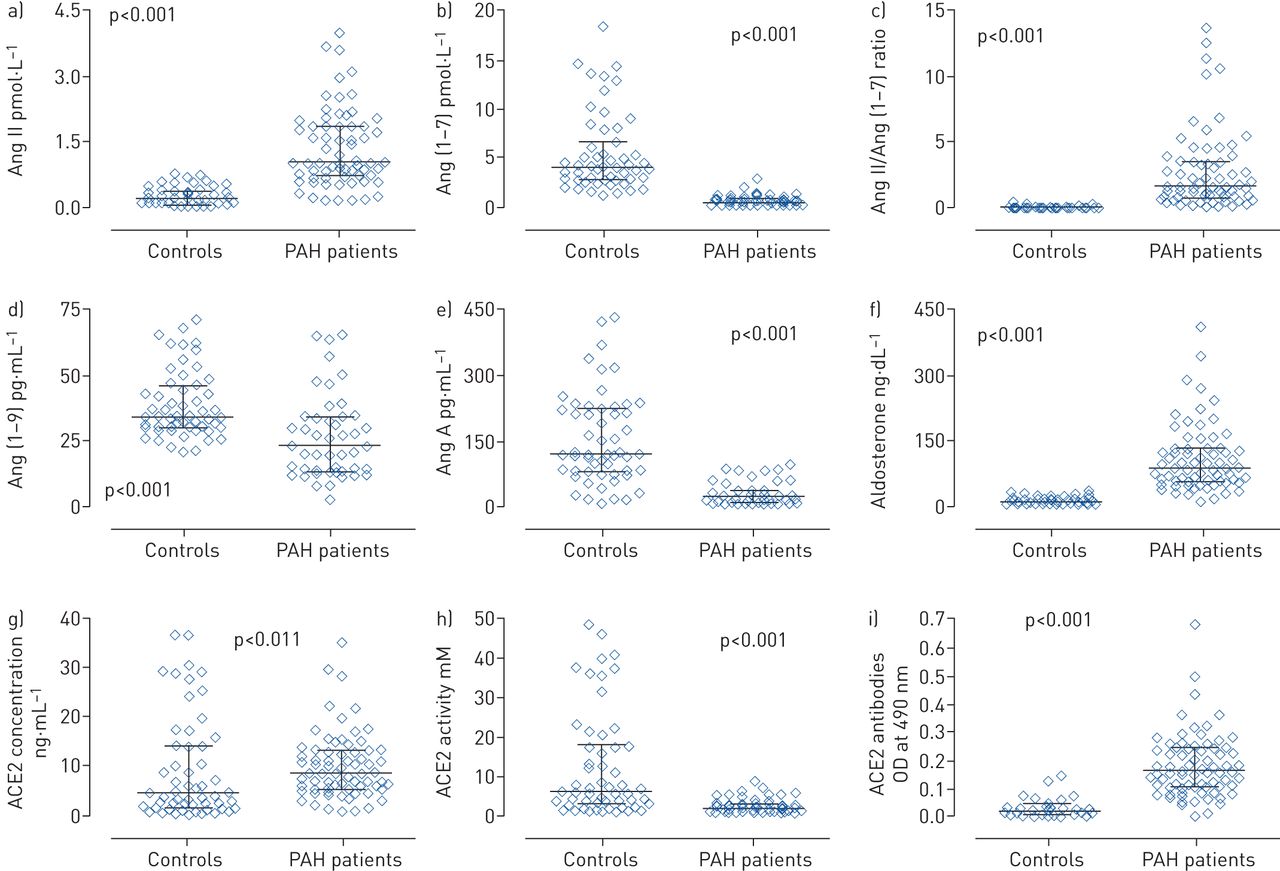
Serum concentrations of AngII, aldosterone, Ang(1–7), Ang(1–9), AngA and ACE 2 levels, as well as ACE2 activity from peripheral vein blood samples were available in 69 PAH patients and the 55 control subjects (table 2 and figure 1). Compared to controls, patients with PAH had a higher concentration of AngII and aldosterone, but a lower concentration of Ang(1–7). Thus, the resulting AngII/Ang(1–7) ratio was significantly higher in PAH patients. In addition, Ang(1–9) and AngA concentration was lower in patients. Unexpectedly, PAH patients had a higher serum concentration of ACE2, while the ACE2 activity was significantly lower than that of controls. In order to explain the diminished activity of ACE2, we searched for serum antibodies directed against ACE2 using a modification of the method described by Takahashi et al. [17] (supplementary material), we demonstrated that anti-ACE2 antibodies not only occur in the three aetiological groups of PAH patients, but the antibody levels are also higher than in healthy controls (table 2).
Peripheral blood serum level measurements of angiotensin (Ang)II, aldosterone, Ang(1–7), Ang(1–9), AngA, angiotensin-converting enzyme (ACE)2 concentration, ACE2 activity and ACE2 antibodies in pulmonary arterial hypertension (PAH) patients and control subjects

Peptide and enzymes differences in patients versus controls. Peripheral serum concentration of a) angiotensin II (AngII) and b) Ang(1–7); c) AngII/Ang(1–7) ratio; peripheral serum concentration of d) Ang(1–9), e) AngA and f) aldosterone; angiotensin-converting enzyme (ACE)2 g) concentration and h) activity; and i) ACE2 antibodies in pulmonary arterial hypertension (PAH) patients and control subjects. OD: optical density. All differences are p<0.001.
Peptide concentrations and enzyme activity differences between PAH subgroups
Serum levels (obtained from the pulmonary artery during RHC) of AngII, Ang(1–7), Ang(1–9) and AngA, and ACE2 concentration as well as ACE2 activity were remarkably similar among the three different subgroups of PAH (table 3). However, the concentration of anti-ACE2 antibodies was higher in PAH-CTD patients (p=0.015) and the anti-ACE2 antibodies correlated negatively with the ACE2 activity only in the IPAH patients (figure 2).
Pulmonary artery blood serum measurements of angiotensin (Ang)II, aldosterone, Ang(1–7), Ang(1–9) and AngA; angiotensin-converting enzyme (ACE)2 concentration, ACE2 activity, and ACE2 antibodies among different pulmonary arterial hypertension (PAH) aetiologies

Correlation between the amount of angiotensin-converting enzyme (ACE)2 antibodies and ACE2 activity in the pulmonary artery blood sample of idiopathic pulmonary arterial hypertension (IPAH) patients. Elevated concentration of antibodies against ACE2 is associated with a decreased ACE2 activity (r=−0.368; p<0.032) in the pulmonary arterial blood sample of IPAH patients. OD: optical density.
As anticipated, for the entire study cohort of PAH patients, the concentration of AngII correlated positively with aldosterone (r=0.331; p=0.002), ACE2 concentration (r=0.607; p<0.001) and ACE2 activity (r=0.377; p<0.001), and negatively with the concentration of Ang(1–7) (r=−0.389; p<0.001). As expected, a higher concentration of ACE2 translated into a similarly higher ACE2 activity (r=0.799; p<0.001).
The levels of Ang(1–7) correlated negatively with the aldosterone concentration (r=−0.256; p=0.020). Lastly, the Ang(1–9) concentration correlated positively with the AngII (r=0.277; p=0.011) and ACE2 concentrations (r=0.227; p=0.039). AngA did not correlate with any other peptide or enzymatic concentrations.
Modifications of enzymes/peptides as a function of passage through the lung
The transpulmonary gradient of enzymes and peptides, as defined by the difference in the concentration between pulmonary artery and systemic (arterial) samples, could be determined in 57 of the PAH patients. There were small, but significant, changes in the enzymes and peptides as they passed through the lung. There was an increase in Ang(1–7) from 0.72 (0.419–0.929) pmol·mL−1 to 0.736 (0.427–0.963) pmol·mL−1 (p<0.001); AngII decreased from 1.186 (0.691–1.909 pmol·mL−1) to 1.158 (0.690–1.928) pmol·mL−1 (p=0.013), and aldosterone decreased from 105.9 (68.7–149.5) ng·dL−1 to 99.7 (67.18–146.9) ng·dL−1 (p=0.020); ACE2 concentration decreased from 8.43 (5.09–13.36) ng·mL−1 to 8.12 (5.00–13.08) ng·mL−1 (p<0.001); and ACE2 activity decreased from 1.79 (1.05–2.48) mm to 1.76 (1.02–2.42) mm (p<0.001). Of interest, in 23 (40.3%) of the patients, aldosterone concentration increased after its passage through the lung circulation (supplementary figure E1).
Correlation between peptide concentrations and enzyme activity with haemodynamics
We found a weak but statistically significant positive correlation between aldosterone concentration and the pulmonary vascular resistance index (r=0.320; p=0.013), as well as a negative correlation with the cardiac index (r=−0.284; p=0.010).
Additionally, we explored the potential clinical importance of the AngII–ACE2–Ang(1–7) axis by analysing its behaviour according to the haemodynamic status of the patients at baseline, as assessed by the cardiac index (table 4). Patients with cardiac index values 2.0–2.5 L·min−1·m−2 had lower values of ACE2 activity and tended to have higher values of aldosterone than patients with a normal cardiac index.
Clinical, functional, haemodynamic, echocardiography and pulmonary artery peptide concentration characteristics of pulmonary arterial hypertension (PAH) patients according to cardiac index
Enzymatic/peptide concentrations in patients with and without ACE2 antibodies
To evaluate the significance of the presence of ACE2 antibodies in PAH patients we determined the cut-off value of antibodies in the PAH patients using the median value of the lower quartile in this population resulting in a median value of 0.076 optical density (OD) at 490 nm (area under the curve=0.937; p<0.001). As noted in table 5, PAH patients with ACE2 antibodies >0.076 OD at 490 nm had a lower ACE2 activity and a higher concentration of aldosterone. No other differences in enzymatic or peptide concentrations were found.
Clinical, functional, haemodynamic, echocardiography and pulmonary artery peptide concentration characteristics of pulmonary arterial hypertension (PAH) patients according to the presence of angiotensin-converting enzyme (ACE)2 antibodies
The impact of previous specific PAH treatments or ACE inhibitors on the axis
52 (61%) out of 85 patients had been treated during different periods (mean (range) 7 (0.5–60) months) with specific PAH drugs (either with sildenafil alone or in combination with an endothelin receptor antagonist or a prostanoid). Except for a higher RAP in the group with previous treatment (6.5 (4–10.2) versus 4 (2–6) mmHg; p=0.016), we did not find any other clinical, haemodynamic or peptide/enzymatic differences between treatment-naïve patients and the group that received drug treatment. We then focused on the 15 treatment-naïve patients (seven with IPAH, five with PAH-CHD and three with PAH-CTD) at baseline and assessed differences in the ACE2–Ang(1–7) axis as a response to PAH-specific pharmacotherapy. These patients were treated with sildenafil alone or combination with bosentan. The results are summarised in supplementary table E1. Despite having improvements in the functional class and 6MWD after a median 7 months of treatment, we did not find any significant change in the peptide concentrations or enzyme activity as measured in the peripheral vein blood.
In addition, we analysed the peptide/enzymatic concentrations in patients with and without medications known to affect the RAAS system, including 26 patients who were taking spironolactone (n=24) with or without ACE inhibitors and angiotensin receptor antagonists or both (n=11), and we compared them with 43 PAH patients not taking these medications and with the 55 control subjects. As shown in supplementary table E2, except for Ang(1–9), all differences in enzymes/peptides are detected between patients (with and without anti-RAAS drugs) and control subjects, but not between patients. However, there are Ang(1–9) differences between patients treated with and without RAAS drugs. It is possible that there is some benefit of these medications in maintaining protective levels of Ang(1–9) in PAH patients.
Peptide concentrations and enzyme activity between pulmonary and peripheral blood
We found no difference in the peptide concentrations or enzyme activity between pulmonary artery blood and peripheral vein blood samples, and their values were highly correlated (supplementary table E3 and figure E2).
Discussion
In this prospective observational study, we demonstrate that, compared with age- and sex-paired healthy controls, patients with PAH have higher levels of AngII, but more importantly, have lower levels of Ang(1–7), Ang(1–9) and AngA. In addition, PAH patients have increased concentrations of ACE2, while its enzymatic activity is decreased compared to healthy controls. We suggest that the reduced activity is in part explained by autoantibodies against ACE2, and that the lower ACE2 activity may explain the lower Ang(1–7) concentrations in PAH patients.
We speculate that increased circulating levels of ACE2 may reflect a compensatory mechanism to alter the balance of the renin angiotensin system in favour of the ACE2-Ang(1–7)-MAS receptor axis and to promote the anti-fibrotic and anti-inflammatory actions of the Ang (1–7), as well as to attenuate the Ang II-AT1 receptor pathway.
Antibodies directed against ACE2 in the serum of idiopathic PAH patients may thus explain the diminished activity of ACE2 (figure 2). Anti-ACE2 antibodies have previously been described in PAH associated with connective tissue disease (CTD) [17]. While in CTD the existence of different types of autoantibodies is not a surprise, we do not have an explanation for the occurrence of anti-ACE2 antibodies in IPAH. Yet our finding supplements the previous description of autoantibodies in IPAH [18] and further supports the hypothesis of autoimmunity in the pathobiology of IPAH [19].
The resultant imbalance in favour of Ang II-aldosterone over ACE2-Ang(1–7) may be an important factor in the pathobiology of PAH. The potential clinical relevance of this finding may be represented by the fact that PAH patients with lower than normal values of CI had lower values of ACE2 activity and higher values of aldosterone than patients with normal CI (table 4).
The RAAS in PAH
The importance of the renin-angiotensin-aldosterone system as a contributor to the pathophysiology of different forms of pulmonary hypertension including idiopathic PAH has been established [8, 9]. It has been shown that certain polymorphisms of ACE and the Angiotensin II receptor 1 (AT1) are associated with disease progression in patients with idiopathic PAH, suggesting that the RAAS is mechanistically involved [20]. High expression of ACE in the endothelium of small pulmonary arteries in the lung tissue from patients with PAH has also been demonstrated [21].
de Man et al. [8] reported that both systemic and pulmonary RAAS activity is increased in patients with IPAH and that these changes are associated with pulmonary vascular remodelling; they demonstrated that in patients with IPAH increased plasma levels of renin, Angiotensin I (AngI), and AngII were closely associated with disease progression and prognosis. They also showed increased ACE activity in isolated pulmonary microvascular endothelial cells, as well as enhanced AngII production after AngI stimulation [8]. AngII also caused increased proliferation of pulmonary artery smooth muscle cells, mediated via enhanced AT1 receptor signalling. Finally, they showed that inhibiting RAAS with losartan may have therapeutic benefits [8]. However, although de Man and co-workers did not explore the counter-regulatory ACE2-Ang(1–7) system, their study demonstrated that the RAAS is indeed activated in patients with IPAH and they provide a rationale for treatment strategies that modify the neuro-hormonal overdrive in these patients [8, 9, 22].
More recently described components of RAAS might also play a role in the pathobiology of PAH. ACE2, which converts AngII to Ang(1–7) is part of a counter-regulatory axis of the RAAS which can affect vasoconstriction, cell proliferation, fibrosis and inflammation [11–14]. In preclinical studies, ACE2 gene transfer prevented the increase in right ventricular (RV) systolic pressure and RV hypertrophy in the monocrotaline rat model and improved RV function in the pulmonary artery banding model [23–25]. Thus, the activation of pulmonary ACE2 probably modifies the pathogenesis of PAH and may serve as a novel therapeutic target in PAH [10, 26, 27].
The ACE2–Ang(1–7) axis in humans with PAH
Previous investigations of the ACE2-Ang(1–7) axis in PAH patients have been limited. Dai et al. [28] reported decreased levels of serum Ang(1–7) in patients with PAH-CHD, and they report a significant negative correlation between the Ang(1–7) levels and mPAP. We could not confirm this result in our study. In the study by Dai et al. [28], Ang(1–7) levels appeared significantly diminished only in CHD patients with severe PAH; the authors did not measure AngII, ACE2 levels, or other components of the axis. Likewise, in a recent proof-of-concept, open-label study on the effect of a soluble recombinant human (rh)ACE2, Hemnes et al. [15] assessed the AngII/Ang(1–7) ratio in 11 patients with idiopathic PAH in order to provide a rationale for the rhACE2 treatment. They showed that the AngII/Ang(1–7) ratio was increased in PAH, but the authors, did not assess other components of the RAAS system.
We considered that ACE2 was also affecting other components of the angiotensin system such as Ang(1–9) or AngA, in addition to Ang(1–7), and measured these peptides in the blood samples. Ang(1–9), another component of the counterbalancing system, is generated from AngI by ACE2, or can be cleaved by ACE to form Ang(1–7) [29, 30]. While little is known about its effects in pulmonary hypertension, preclinical studies [31] have demonstrated that Ang(1–9) reduced RV systolic pressure and RV hypertrophy and attenuated endothelial damage and medial hypertrophy of pulmonary arterioles as well as pulmonary fibrosis induced by monocrotaline. Ang(1–9) inhibited the infiltration of inflammatory cells, reduced the pro-inflammatory cytokines and attenuated expression of apoptosis-related proteins. All these pulmonary vascular disease-modifying effects of Ang(1–9) are being signalled through the angiotensin type 2 (AT2) receptor.
The concentration of Ang(1–9) in our PAH patients was lower than that in control subjects (table 2); this is consistent with the lower ACE2 activity. As mentioned, Ang(1–9) can be cleaved by ACE to form Ang(1–7) [29–31], and thus it is not possible to establish here whether the low levels of Ang(1–7) in our patients are a consequence of a low concentration of Ang(1–9). Whatever the mechanism, our results indicate that, in a similar way to the ACE2/Ang(1–7)/Mas receptor, the ACE2/Ang(1–9)/AT2 receptor axis, another counter-regulatory arm of RAAS to ACE/Ang II/angiotensin type 1 (AT1) receptor, is also dysfunctional in PAH (figure 3).

{kind=link}
{kind=link}
{kind=link}
Schematic representation of the renin–angiotensin–aldosterone system (RAAS) and its counterbalance in pulmonary arterial hypertension (PAH), according to our findings. In patients with PAH, several components of the counterbalancing axis of RAAS (angiotensin (Ang)(1–7), Ang(1–9) and AngA–alamandine) are affected and probably impaired. Although serum levels of angiotensin-converting enzyme (ACE)2 are elevated, its activity is diminished (#), perhaps in part due to autoantibodies against ACE2. Renin, AngI, ACE and peptide receptors were not measured. APA: aminopeptidase A; APM: aminopeptidase M; AT1: angiotensin type-1 receptor; AT2: angiotensin type-2 receptor; AT4: angiotensin type-4 receptor; Mas: Mas receptor; MrgD: Mas-related G protein coupled receptor; MLDAD: mononuclear leukocyte-derived aspartate decarboxylase. Reproduced and modified with permission [40].
The concentration of AngA was diminished in our PAH patients as compared to normal controls (table 2). Its role in the setting of PAH remains uncertain. AngA may elicit either direct vasoconstrictive and pro-proliferative actions via the AT1 receptor [32], or it can be further metabolised to alamandine (hydrolysed by ACE2 from AngA), triggering opposing effects via the Mas-related G protein coupled receptor (MrgD) receptor. Alamandine can be synthesised from Ang(1–7) by decarboxylation of aspartate to form alanine. Accordingly, alamandine, a central molecule of this counter-regulatory cascade, can be generated both from AngA as well as from Ang(1–7) [32]. As these peptides, Ang(1–7) and alamandine, have similar amino acid sequence and structure, their effects are highly likely to be similar. Their respective receptors (Mas receptor (Ang(1–7)) and MrgD receptor (alamandine)) are expressed on endothelial cells and they may play important roles in vascular physiology [32].
Taken together, our results suggest that in patients with PAH, several components of the counterbalancing axis of RAAS (Ang(1–7), Ang(1–9) and AngA–alamandine) are affected and probably impaired. A schematic representation of our findings is depicted in figure 3. The diminished ACE2 activity in this protective mediator cascade is probably of central importance, giving support to current studies targeting the downstream ACE2/Ang(1–7)/Mas receptor pathway. It is interesting that the components of the ACE2–Ang(1–7) axis, including Ang(1–9) and AngA are remarkably similar among the three different subgroups of PAH despite the clinical and haemodynamic differences between them, a finding which may be explained by a possible shared endothelial cell pathobiology. It is also of interest that the peptide/enzymatic activity values were not altered in the patients following targeted therapy for PAH: we had initially postulated that the drug treatment would normalise these peptide values. The finding of an increased level of Ang(1–9) in patients taking drugs that modify the RAAS (supplementary table E2) is interesting and deserves further investigation.
Implications for treatment
Therapeutic modification of the neurohormonal axis in patients with severe PAH appear to be possible and the search for additional therapies for this catastrophic group of diseases is warranted. There is evidence for the beneficial effect of modifying conventional components of the RAAS [8–10, 22]. Most importantly, there is now evidence for the potential benefit of modifying the ACE2–Ang(1–7) axis in humans with PAH. Hemnes et al. [15] found that a single infusion of rhACE2, a purified intravenous formulation of soluble recombinant human ACE2 (GSK2586881) was well tolerated and was associated with improved pulmonary haemodynamics and reduced marker levels of oxidant and inflammatory stress. The results of our large cohort study strengthen the rationale for such a therapeutic approach by confirming that in human PAH the ACE2–Ang(1–7) axis is indeed abnormally expressed. In addition, our data suggest that increasing the level of ACE2 may be ineffective when ACE2 is not functional. An effective strategy might be to assure ACE2 functionality, to increase the levels of Ang(1–7) and to assure the proper functioning of its MAS receptor. Removal of the ACE2 antibodies, for example by means of plasmapheresis, or perhaps by recombinant antibodies that neutralise the action of ACE2 antibodies, may optimise the therapeutic benefits of ACE2 agonists/activators.
Another important finding of our study was the confirmation of a disproportionate increase of aldosterone in PAH patients and that high aldosterone levels inversely correlated with the cardiac index (supplementary figure E3). Preclinical and clinical studies have demonstrated that AngII and aldosterone regulate cellular signalling processes that increase cell proliferation, migration and vascular hypertrophy, as well as initiate cardiovascular fibrosis and interrupt repair processes [33]. Indeed, animal models of PAH have demonstrated the beneficial effects of mineralocorticoid receptor antagonism with spironolactone or eplerenone [26, 27]. In the clinical setting, the addition of spironolactone to targeted treatment with ambrisentan in the ARIES studies [26, 34] has suggested some clinical benefit as well. The precise role of these drugs remains to be studied in randomised clinical trials.
Study limitations
First, our study does not have validation cohort; therefore, our results must be confirmed in another study within a different population and with a larger sample size. Second, although our analysis did not show significant differences in patients with and without PAH treatment, we cannot exclude completely other PAH-drug influences in our results, as this was not a prospectively designed intervention in our study. Third, our patients were diagnosed at a relatively late stage of the disease and some of the aetiology groups (PAH-CTD) were relatively small; accordingly, further information on early-stage disease and aetiology remains to be elucidated. Finally, in the three different groups of patients we studied, inflammation probably plays a role. Accordingly, biomarkers of inflammation should be assessed/monitored in future studies.
Conclusions
In patients with idiopathic, CTD- and CHD-associated PAH there are significant abnormalities in the AngII–ACE2–Ang(1–7) axis characterised by elevated levels of AngII and aldosterone. Decreased Ang(1–7), Ang(1–9) and AngA levels are perhaps explained by decreased ACE2 activity. Although serum levels of ACE2 are elevated, its activity is diminished, probably at least in part due to autoantibodies against ACE2. In addition, we found elevated concentrations of aldosterone. Elevated blood levels of aldosterone have been reported in patients with left heart failure [35], and it has been proposed that in pulmonary hypertension extra-adrenal lung vascular endothelial cells produce aldosterone [33] which may stimulate vascular smooth muscle cell growth in a paracrine fashion [36]. As shown in supplementary figure E1, aldosterone concentration increased after its passage through the lung in a significant proportion of our patients, but not in all patients. These findings are an illustration of the endothelial cell phenotypical alterations, which characterise the “sick lung circulation” of PAH patients [37].
Our results not only show reduced ACE2 activity, but also illustrate how AngII may preferentially signal via the angiotensin 1 and/or 4 receptor, shifting the balance towards vasoconstriction, cell proliferation, inflammation and fibrosis, thus contributing, together with aldosterone [33], to the pathobiology of PAH.
In a different and highly relevant setting, it is also of interest that ACE2 is the cellular receptor for coronaviruses [38]. Variable expression of ACE2, for example due to sex-associated polymorphisms, may influence the pulmonary inflammatory response and possibly play a role in the development of pulmonary vascular diseases, as ACE2 has been shown to modify angiogenesis via inhibition of vascular endothelial growth factor receptor 2 signalling [39].
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02416-2019.SUPPLEMENT
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02416-2019.Shareable
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: Conception or design: J. Sandoval, F. Masso, G. Pastelin-Hernandez, J. Gómez-Arroyo and N.F. Voelkel. Acquisition, analysis, interpretation of data: J. Sandoval, L. Del Valle-Mondragón, F. Masso, N. Zayas, T. Pulido, R. Teijeiro, H. González-Pacheco, R. Olmedo-Ocampo, C. Sisniega and A. Paez-Arenas. Drafting or revising the work: J. Sandoval, J. Gómez-Arroyo and N.F. Voelkel.
Conflict of interest: J. Sandoval has nothing to disclose.
Conflict of interest: L. Del Valle-Mondragón has nothing to disclose.
Conflict of interest: F. Masso has nothing to disclose.
Conflict of interest: N. Zayas has nothing to disclose.
Conflict of interest: T. Pulido reports grants from and personal fees for advisory board work and lectures from Actelion and Bayer, grants from Lilly, Reata Pharmaceuticals and United Therapeutics, personal fees for advisory board work from Pfizer and Akros Pharma, outside the submitted work.
Conflict of interest: R. Teijeiro reports grants from CONACYT (FOSISS project 2015-1-262511), during the conduct of the study.
Conflict of interest: H. González-Pacheco has nothing to disclose.
Conflict of interest: R. Olmedo-Ocampo has nothing to disclose.
Conflict of interest: C. Sisniega has nothing to disclose.
Conflict of interest: A. Paez-Arenas has nothing to disclose.
Conflict of interest: G. Pastelin-Hernandez has nothing to disclose.
Conflict of interest: J. Gómez-Arroyo has nothing to disclose.
Conflict of interest: N.F. Voelkel has nothing to disclose.
Support statement: This work was supported by CONACYT (FOSISS project 2015-1-262511). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received November 8, 2019.
- Accepted March 21, 2020.
- Copyright ©ERS 2020
References










