





Abstract
Systemic sclerosis (SSc) is a systemic autoimmune disease affecting multiple organ systems, including the lungs. Interstitial lung disease (ILD) is the leading cause of death in SSc.
There are no valid biomarkers to predict the occurrence of SSc-ILD, although auto-antibodies against anti-topoisomerase I and several inflammatory markers are candidate biomarkers that need further evaluation. Chest auscultation, presence of shortness of breath and pulmonary function testing are important diagnostic tools, but lack sensitivity to detect early ILD. Baseline screening with high-resolution computed tomography (HRCT) is therefore necessary to confirm an SSc-ILD diagnosis. Once diagnosed with SSc-ILD, patients' clinical courses are variable and difficult to predict, although certain patient characteristics and biomarkers are associated with disease progression. It is important to monitor patients with SSc-ILD for signs of disease progression, although there is no consensus about which diagnostic tools to use or how often monitoring should occur. In this article, we review methods used to define and predict disease progression in SSc-ILD.
There is no valid definition of SSc-ILD disease progression, but we suggest that either a decline in forced vital capacity (FVC) from baseline of ≥10%, or a decline in FVC of 5–9% in association with a decline in diffusing capacity of the lung for carbon monoxide of ≥15% represents progression. An increase in the radiographic extent of ILD on HRCT imaging would also signify progression. A time period of 1–2 years is generally used for this definition, but a decline over a longer time period may also reflect clinically relevant disease progression.
Abstract
Lung function tests and chest imaging help predict who has SSc-associated ILD and whether it will progress. In the absence of standardised methods for doctors, we recommend a strategy that combines both lung function tests and chest imaging. http://bit.ly/2uK9ZD2
Introduction
Systemic sclerosis (SSc) is a systemic autoimmune disease that is characterised by endothelial dysfunction, resulting in small-vessel vasculopathy, immune dysregulation, fibroblast dysfunction and subsequent fibrosis; however, its detailed pathogenesis remains unclear [1]. Due to the heterogeneity of the disease, SSc represents a major clinical challenge for both physicians and patients [2]. In addition to disfiguring skin involvement, SSc can affect multiple organ systems, including the lungs [3]. The clinical course is variable, but organ manifestations frequently occur early in the disease [4]. SSc is associated with a 250% increase in mortality risk compared with healthy controls and, overall, this has not changed significantly in the past 40 years [5]. Mortality is primarily due to pulmonary complications: in the largest observational study conducted to date, the leading cause of death was interstitial lung disease (ILD; 17%), with pulmonary arterial hypertension (PAH) accounting for 15% of deaths [6, 7]. Furthermore, in a Norwegian cohort study, mortality in SSc correlated with the extent of lung fibrosis [8].
Between 1972 and 2002, the proportion of deaths due to scleroderma renal crisis declined from 42% to 6% of SSc-related deaths, probably due to the identification of risk factors, prevention, the use of angiotensin-converting enzyme inhibitors to treat this dimension of the disease, and a greater awareness of milder SSc cases less likely to develop renal crisis. In contrast, over the same period, the proportion of SSc deaths due to ILD increased from 6% to 33% [9], and in recent interventional studies, respiratory failure due to ILD was reported to account for 43% of deaths [10]. This increase in the proportion of deaths due to ILD may reflect decreases in other causes of deaths. The overall 10-year survival improved from 54% to 66% from 1972 to 2002 [9].
Patients with SSc are routinely divided into limited cutaneous (lcSSc) and diffuse cutaneous (dcSSc) subsets based on the extent of skin fibrosis [11, 12]. Pulmonary involvement can occur in both subsets of the disease, and it can affect all aspects of the respiratory tract, including the parenchyma, vasculature and musculature [8, 13]. ILD is an early complication in SSc, and in some patients (∼4%) the first clinical symptom of SSc is directly related to ILD [2, 14]. Most patients who develop severe restrictive lung disease do so in the first 5 years following the onset of SSc symptoms [2].
Despite the established relationship between SSc-associated ILD (SSc-ILD) and morbidity and mortality, there is still no consensus on screening for ILD, nor on monitoring for disease progression. This issue is further complicated by the lack of validated biomarkers for SSc-ILD and an absence of clinical recommendations to inform the method and timing of investigations to diagnose patients with SSc-ILD and identify those at risk of progression [15].
In this perspective piece, we summarise the current understanding of disease pathogenesis and risk factors for the presence of ILD in patients with SSc, discuss screening, early detection of ILD and risk factors for progression, and propose guidance on monitoring disease progression and the implications of this for treating SSc-ILD.
Pathogenesis of SSc-ILD
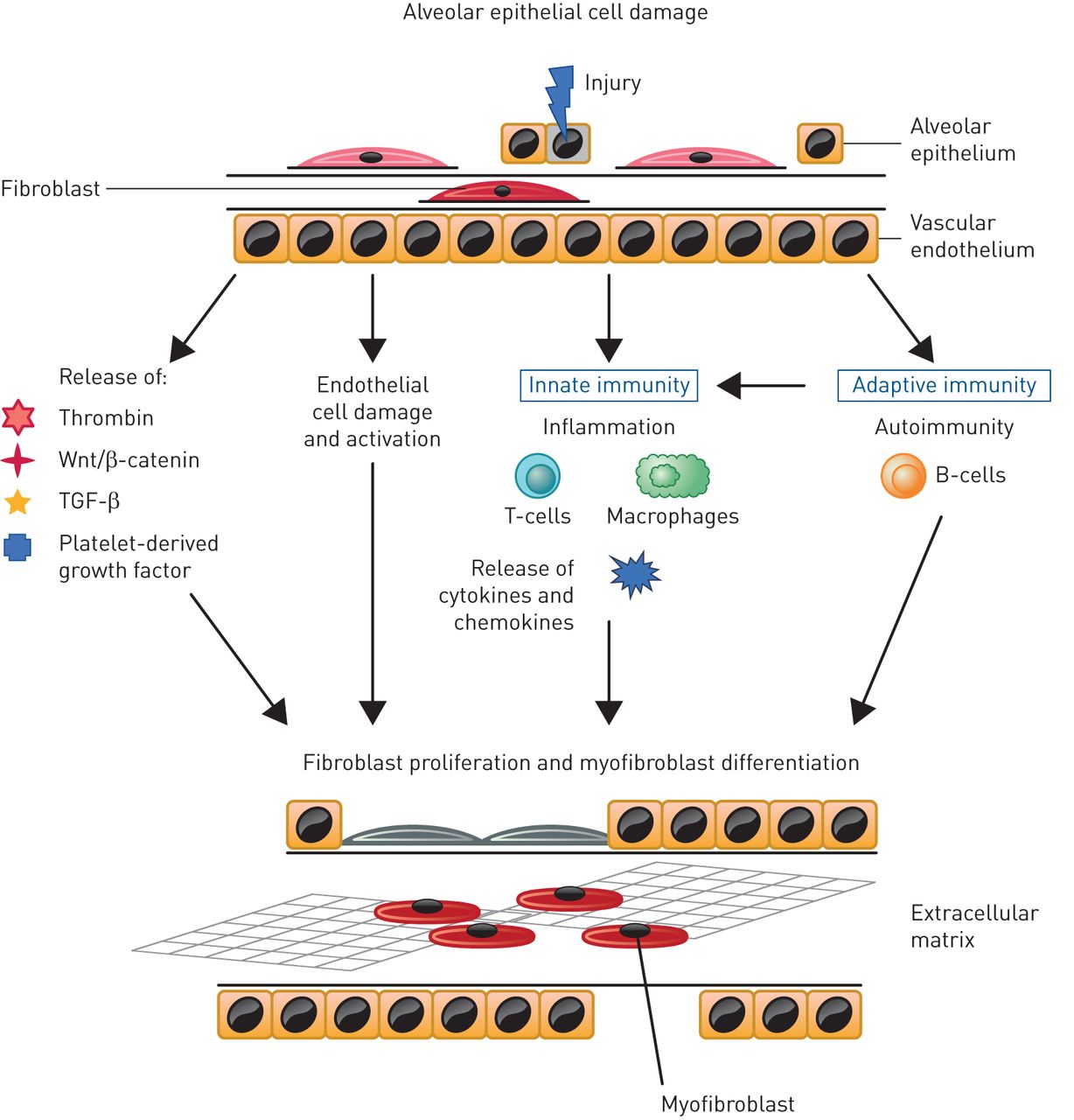
SSc may affect multiple organs and systems, with most irreversible damage attributable to progressive fibrosis [2, 16]. Based on research to date, it is likely that there is a complex interplay between innate and adaptive immunity, endothelial dysfunction, small vessel abnormalities, inflammation and fibrosis [17]. The key pathways implicated in SSc-ILD are summarised in figure 1. The most common initiator of the pathogenesis of SSc-ILD is injury to alveolar epithelial and vascular endothelial cells, in which inflammatory pathways lead to profibrotic stimuli that induce the differentiation of lung fibroblasts to the myofibroblast phenotype [6, 18]. Fibrosis results from an abnormal healing process coupled with an aberrant expansion of mesenchymal cells that originate from multiple sources including bone marrow, the transdifferentiation of pericytes, epithelial cells and endothelial cells, and the differentiation from resident fibroblasts to myofibroblasts [16, 18]. Transforming growth factor-β is thought to play a central role in the development and progression of fibrosis in SSc-ILD [6, 16] and platelet-derived growth factor, Wnt-β-catenin and hedgehog signalling also drive fibrosis [18, 19]. SSc fibroblasts have been shown to be more resistant to the fibrotic-inhibitory potential of cytokines, such as interferon-γ [20]. Furthermore, patients with SSc-ILD can have increased levels of pro-inflammatory cytokines such as interleukin (IL)-8, IL-6, tumour necrosis factor-α and macrophage inflammatory protein-1α in bronchoalveolar lavage (BAL) fluid and serum [21–23]. In addition, it has been hypothesised that B-cells may be involved, as patients with SSc-ILD more frequently present with anti-topoisomerase I antibodies and antifibroblast antibodies [24]. The functional role of antifibroblast antibodies has not been clarified, but they might directly affect extracellular matrix remodelling by binding to fibroblasts to increase the collagen breakdown and matrix metalloproteinase-1 production [25]. The increased release of these cytokines, growth factors and bioactive proteins contribute to a self-perpetuating cycle of stimulation of the fibrotic process [16, 18]. A greater understanding of the underlying mechanisms may enable the identification of biomarkers that could be used to screen at-risk patients and facilitate the development of targeted and personalised therapy. For instance, higher-circulating mixed M1 (classically activated)/M2 (alternatively activated) monocyte/macrophage cell percentages are associated with ILD, suggesting a need for research on their possible role as a biomarker or pathogenic element for SSc lung involvement [26].

Risk factors for presence of ILD
Estimates of the prevalence of ILD in SSc vary depending on how ILD is defined; using high-resolution computed tomography (HRCT), prevalence is estimated at 47–84%. In the Canadian Scleroderma Research Group registry, 65% of patients had evidence of ILD on HRCT scans, 26% had ILD identified by physical examination and 22% identified by chest radiography [27]. In a Norwegian cohort, 50% of patients had ILD on HRCT [8]. Relying on pulmonary function testing (forced vital capacity (FVC) % predicted or a decrease in at least one pulmonary function test (PFT)) results in a lower estimate of prevalence of ILD [28]. At autopsy, evidence of interstitial fibrosis was found in 74% of patients with SSc [29].
A number of patient characteristics are associated with a higher risk of developing ILD in SSc (table 1). In the EUSTAR (EULAR Scleroderma Trials and Research) database, ILD was more common in dcSSc (53.4%) than in lcSSc (34.7%) [30]. In addition, sex and ethnicity appear to be important determinants of ILD development. Despite the strong female predominance reported in SSc (female:male ratio 4.7:1), males with SSc are more likely to develop ILD than females with SSc (relative risk 1.24; 95% CI 1.01–1.52) [31, 32]. A large multi-ethnic observational study conducted in the setting of a universal healthcare system found that in comparison to white patients of European descent, patients of Afro-Caribbean descent with SSc more frequently have ILD (31% versus 53%; p=0.007) [33].
Clinical and biological factors associated with the presence of interstitial lung disease in systemic sclerosis
Risk factors for the development of SSc-ILD also include anti-topoisomerase I positivity, lower FVC and lower diffusing capacity of the lung for carbon monoxide (DLCO) [34], although FVC and DLCO changes may be caused by ILD and not present before ILD onset. Multivariate analyses suggest that the risk of developing ILD is more closely associated with anti-topoisomerase I positivity than to dcSSc versus lcSSc [30].
A number of studies also support an association of gastroesophageal reflux with ILD in SSc, as reviewed by Christmann et al. [35].
Diagnosis and screening of ILD
The most common initial symptoms of SSc-ILD are exertional dyspnoea and dry cough, although early SSc-ILD can be relatively asymptomatic [6]. As lung involvement can affect all aspects of the respiratory tract, when a patient with SSc presents with symptoms of dyspnoea, the differential diagnosis can be quite broad and may include PAH, deconditioning, chronic anaemia [13] and left heart involvement with reduced or preserved ejection fraction.
SSc-ILD is diagnosed using HRCT to detect parenchymal lung disease [36]. Typically, a pattern suggestive of nonspecific interstitial pneumonia (NSIP) appears on HRCT as bilateral ground-glass opacities, reticulation and traction bronchiectasis most prominent in the lower lobes (figure 2a) [36]. Generally, HRCT appearance can differentiate NSIP and usual interstitial pneumonia (UIP) patterns [37]. Although lung biopsy is very rarely indicated to diagnose SSc-ILD, if performed, interstitial fibrosis with temporal homogeneity and a modest inflammatory cell infiltrate (i.e. fibrotic NSIP) is typically visualised (figure 2b). In a small number of cases, cellular NSIP or UIP are observed [13]. Although computed tomography (CT) scans are necessary for diagnosis, they expose patients to radiation [38]. Reduced-dose HRCT protocols may reliably detect SSc-ILD with a much lower radiation exposure than whole-chest HRCT [38, 39]. While these reduced-dose protocols have become routine in some centres, further validation in larger cohorts is required.

a) High-resolution computed tomography from a patient with systemic sclerosis (SSc)-associated interstitial lung disease (disease extent >20%); b) lung histology from a patient with SSc showing nonspecific interstitial pneumonia.
PFTs in patients with SSc-ILD generally show a restrictive ventilatory defect with a decreased FVC and/or total lung capacity, a preserved forced expiratory volume in 1 s (FEV1), and a normal or increased FEV1/FVC ratio, as well as a decreased single-breath DLCO [6]. It is important to note that patients with early SSc-ILD may have preserved lung volumes, despite clear evidence of structural lung disease on HRCT [28, 40], and decreases in DLCO. Two studies found that >60% of patients with SSc and ILD diagnosed using HRCT had normal spirometry [28, 40]. Therefore, while PFTs are an important part of the diagnostic work-up for SSc-ILD, they are not sensitive enough to reliably detect early ILD [28, 40, 41]. Performing PFTs regularly (at least once a year) in the first 5 years following SSc diagnosis may be useful for detecting changes in lung function indicative of ILD.
At present, there is no widely accepted consensus on when SSc-ILD becomes clinically significant and when treatment is indicated.
Challenges in diagnosis
There are many challenges associated with the diagnosis of SSc-ILD. One potential barrier to diagnosis is a lack of awareness within primary care of SSc, which can lead to late referrals [42]. There is also variability in the degree of involvement of clinical specialties in establishing a diagnosis of SSc-ILD, and a lack of formally designated SSc specialist centres [42].
Early detection of ILD
There is a need for early systematic screening of patients to identify pulmonary complications, although there is a lack of consensus regarding which tests should be used during screening and follow-up [2, 42]. In one study, screening for ILD at diagnosis of SSc using HRCT identified 35% of subjects with no ILD, 51% of subjects with ILD of limited extent (1–20% fibrosis) and 13% of subjects with ILD involving >20% of the lungs [43].
Subclinical interstitial lung abnormalities (ILAs) detectable on CT scans offer a possible approach to detect ILD early [44, 45]. ILAs have been studied in rheumatoid arthritis-associated ILD, where they are associated with increased cough and dyspnoea and decreased PFT results [46]; however, there is little evidence characterising them in SSc-ILD. One study in early SSc-ILD demonstrated ground-glass opacities that later progressed to NSIP pattern [47].
Future directions in the diagnosis and screening of SSc-ILD
In future, lung ultrasound may be integrated with other approaches in the screening of ILD [48–51]. Preclinical studies of positron emission tomography scanning suggest that targeted nuclear imaging of components of ILD disease processes (such as integrin αvβ3 and somatostatin receptor 2) could be integrated with other approaches in the screening and evaluation of ILD [52, 53]. However, this method is limited by the high levels of radiation exposure associated with the technique.
Another potential approach to help identify patients with ILD is nailfold videocapillaroscopy (NVC). NVC is a safe and validated tool, and NVC patterns are associated with different SSc clinical complications [54, 55]. NVC was tested in 287 patients with SSc to evaluate whether it could improve the detection of patients at high risk of cardiopulmonary involvement. In this analysis, NVC pattern was associated with the presence of ILD and heart/lung involvement, independent of specific anti-extractable nuclear antigen antibodies [56]. This might indicate that microangiopathy is an important cause of organ involvement. In future, NVC may be incorporated, together with other parameters, in high-performance algorithms in the early detection of lung involvement in SSc [57].
However, despite identified risk factors for the development of ILD in patients with SSc, and newer techniques that may contribute to identify subpopulations with increased risk of developing ILD, currently none of the available techniques other than CT can detect ILD with appropriate sensitivity and specificity. Therefore, chest CT remains the gold standard to screen for and diagnose SSc-ILD.
Risk factors for progression of ILD
Once ILD has been diagnosed in patients with SSc, it is important to identify which patients are likely to progress, because the natural history of SSc-ILD is variable. Some patients experience accelerated loss of lung function, and others progress slowly or exhibit stable disease [58, 59].
At present, there is no biomarker that predicts the course of SSc-ILD across multiple clinical cohorts [60]. Initial reports that BAL cellularity could predict mortality, benefit of immunosuppressive therapy, progression or progression-free survival [61, 62] have not been substantiated in studies examining larger cohorts [63]. However, several strategies are available to help identify patients at risk for the development and progression of SSc-ILD, and these should be considered as a first step in the management of patients with SSc (table 2).
Clinical and biological factors associated with progression of systemic sclerosis-associated interstitial lung disease (ILD)#
In clinical practice, the severity of SSc-ILD may be staged based on the “Goh criteria”, whereby patients have “limited” or “extensive” disease based on HRCT (clearly <20% or clearly >20%, respectively), with the use of an FVC threshold of 70% in indeterminate cases [41]. Baseline HRCT extent has been associated with decline in FVC % pred [64] and, in a more recent study, extensive disease (>20%) was associated with a threefold increase in deterioration or death [65].
An evidence-based prediction model that combines peripheral capillary oxygen saturation (SpO2) and arthritis history (SPAR) has been developed to predict progression of mild ILD [66]. In 215 patients with SSc and mild ILD, lower SpO2 after a 6-min walk test and the presence of arthritis at any point were independent predictors of ILD progression (defined as a relative decrease in FVC ≥15%, or FVC ≥10% combined with DLCO ≥15% at 1-year follow-up) [66]. By combining both predictors, the derived SPAR model was able to increase the prediction rate from 25.5% to 91.7% and, ultimately, may enable risk stratification of patients with early, mild SSc-ILD [66].
A number of other risk factors for SSc-ILD disease progression have been identified. A large SSc-ILD cohort study demonstrated that FVC (hazard ratio (HR) 0.97; p=0.0009), DLCO (HR 0.97; p=0.0004) and increasing age (HR 1.03; p=0.04) were significantly associated with mortality after adjusting for sex, body mass index, smoking status, serology, disease subtype and immunosuppressive use [67]. A recent analysis showed that a decline in FVC or DLCO over 2 years was a better predictor of mortality than baseline FVC and DLCO [10].
Auto-antibodies against the DNA-modifying enzyme topoisomerase I are frequently present in patients with dcSSc and may be associated with the faster progression of SSc-ILD [34, 68, 69]. Owing to the pathogenic role of the chemokine receptors CXCR3 and CXCR4 in fibrosis, auto-antibodies against them have been investigated in patients with SSc [70]. Anti-CXCR3 and anti-CXCR4 antibody levels have been shown to be different among SSc subgroups compared with healthy subjects, and highest in diffuse SSc patients. Patients with SSc-ILD exhibited higher antibody levels than those without ILD; however, patients with deterioration of lung function (FVC <70% pred) showed lower anti-CXCR3/4 antibody levels compared with those with stable disease [70]. However, these biomarkers are not routinely available.
Several inflammatory markers have been investigated. Elevated serum chemokine ligand 18 is predictive of FVC decline [71, 72] and is a risk factor for death or progression of SSc-ILD [72–74]. Baseline C-reactive protein levels predict long-term progression of SSc-ILD, and higher levels are associated with shorter survival [75] and a decline in PFTs [76]. Krebs von den Lungen (KL)-6 and surfactant protein-D are proteins predominantly expressed in the lung, and are associated with lung damage, fibrosis and inflammation [72, 77]. Serum KL-6 level is inversely correlated with DLCO % pred, and elevated levels are associated with end-stage lung disease and faster FVC decline [72, 77–79], even in the presence of treatment with immunosuppression [80]. Further research is required to validate these biomarkers.
Monitoring disease progression
The variable rates of disease progression in SSc-ILD mean that it is important that patients are appropriately monitored after a diagnosis of ILD in SSc, although there is currently no established definition for disease progression. Serial measurements of symptoms (e.g. appearance or aggravation of dyspnoea, cough and/or fatigue), physical examination and PFTs are important to assess disease progression at regular clinic visits [6, 21], and recent studies highlight the impact of change in PFT on mortality [10, 81].
In addition, patient-reported outcomes may be important for measuring disease progression; there is an association between lower FVC and worsening scores on St George's Respiratory Questionnaire (SGRQ), a measure of respiratory-related health status, in patients with ILD [82]. It should be noted that this study largely evaluated patients with idiopathic pulmonary fibrosis, hypersensitivity pneumonitis and NSIP; SGRQ scores in SSc may be affected by comorbidities, including musculoskeletal problems.
Outstanding questions remain regarding the use of repeated HRCT, including how often it should be performed routinely (for instance, every year or every other year), and whether PFT or other tools should be used for monitoring, with HRCT only used if necessary (e.g. unexpected worsening or suspicion of infection) [38, 83]. In addition, there is some debate as to whether and how often CT scans should be repeated in patients with SSc who do not have an ILD when first examined.
SSc-ILD expert panel
An international panel of SSc-ILD experts from Europe and North America was assembled to discuss developments in the field. Topics of interest were selected by panel chairs and discussed in groups. The selected group of pulmonologists and rheumatologists identified predictors of progression of SSc-ILD as an area of unmet need. We reviewed evidence for the clinical course of SSc-ILD, examined the challenges involved in diagnosis, and provided guidance on the staging and risk stratification of patients with SSc-ILD.
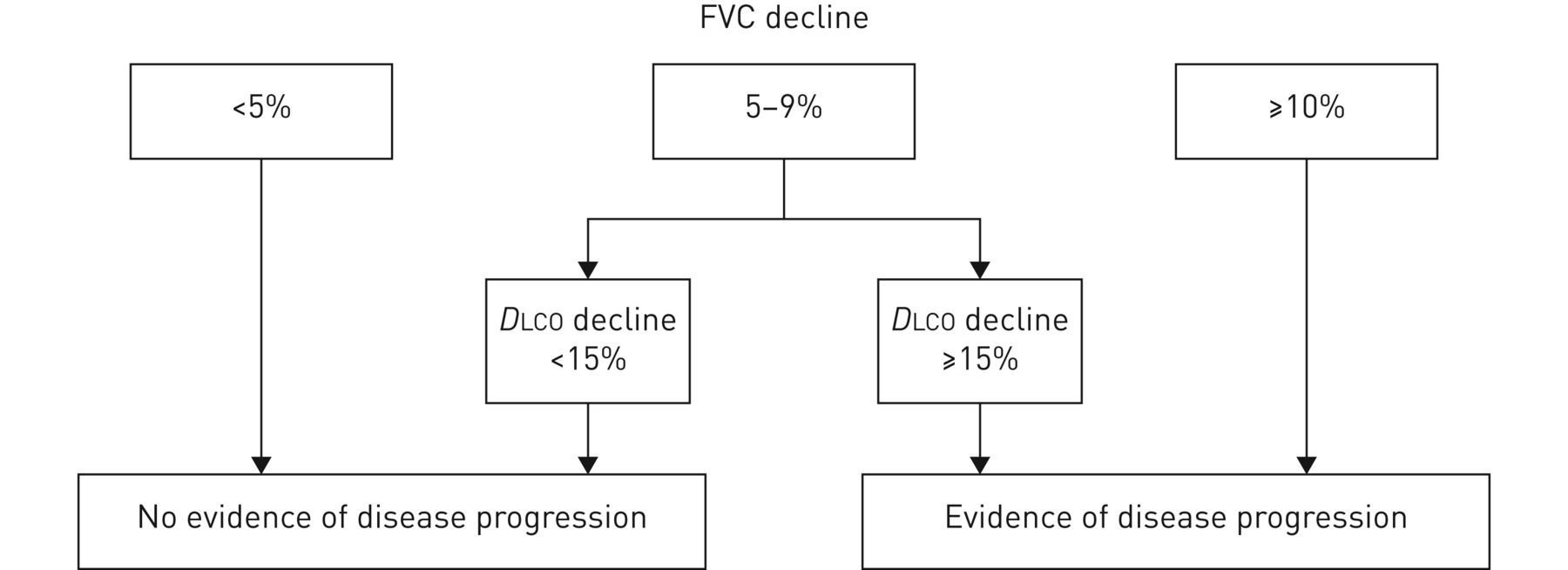
We support the definition of progression of disease in individual patients as a sustained change in lung function characterised by an FVC decline from baseline of ≥10%, or an FVC decline of 5–9% in association with a DLCO decline of ≥15% (figure 3) [81]. If serial CTs were to show an increase in the extent of ILD on HRCT imaging this would also be taken into account. This definition is one of the most commonly used definitions for progression, although it has not been formally validated in SSc-ILD. Researchers usually use this definition with a time period of 1 year, but there is no agreement on how disease progression should be defined over a longer time period. It should be acknowledged that this decline in FVC and/or DLCO observed over a longer time period still demonstrates potentially clinically relevant disease progression, albeit at a slower rate.

{kind=link}
{kind=link}
{kind=link}
Proposed definition of disease progression. FVC: forced vital capacity; DLCO: diffusing capacity of the lungs for carbon monoxide.
Without intervention, the natural annual loss of lung function is 15–25 mL (inferred from regression equations in [84]). Diurnal variability in lung function is 8–9% and measurement error in clinical trials is ∼1% [85]. In the worst-case scenario, this amounts to 10%. Thus, for a relative 5–9% loss in FVC, we agreed that it should be accompanied by a decline in DLCO to confirm disease progression.
Implications for therapy
In the recently published Safety and Efficacy of Nintedanib in Systemic Sclerosis (SENSCIS) trial, patients with SSc-ILD treated with nintedanib had a lower rate of annual FVC decline than those receiving placebo (treatment difference 41 mL) [86]. Based on these data, nintedanib was recently approved by the US Food and Drug Administration for slowing the rate of decline in pulmonary function in patients with SSc-ILD [87]. Guidelines issued by the European League Against Rheumatism and the EUSTAR group in 2017 recommend consideration of tailored cyclophosphamide (CYC) treatment in patients with SSc-ILD, and the use of haematopoietic stem cell transplant in selected patients with rapidly progressing SSc [88]. Following the Scleroderma Lung Studies (SLS) II trial [10], a Delphi consensus treatment algorithm advocated for first-line treatment of SSc-ILD with mycophenolate mofetil (MMF). Second-line treatments included CYC or rituximab as induction therapy, followed by MMF as maintenance therapy [89].
However, there is currently no consensus regarding treatment initiation or escalation [90]. Most SSc experts draw on their clinical experience and consider the risk factors for SSc-ILD progression to inform their decision on when to initiate therapy on a case-by-case basis [91]. Better identification of patients at greatest risk of progression will help to identify those who can benefit the most from early treatment [6]. There is a clear need for new treatments. As our understanding of the pathogenesis of SSc-ILD evolves, there is the hope that specific therapies will be introduced that treat the disease in the organ systems affected, improving patient wellbeing, function and survival [92–95]. Targeting the self-perpetuating fibrosis may provide therapeutic options in the future [16], although it remains unknown whether early aggressive treatment of the disease is beneficial in terms of modifying disease progression and preventing irreversible lung damage. In the near future, it is hoped that molecular detection of disease will inform immunomodulatory therapy in SSc patients [96].
Conclusions
By increasing awareness among physicians of frequency, progression and mortality associated with SSc-ILD, there is the potential to improve patient outcomes. Better identification of which patients are at risk of progression will also be beneficial. ILD is an early complication of SSc, and there is a need for early and systematic screening of patients to potentially enable treatment prior to deterioration of lung function in subjects at high risk of disease progression. Pulmonary function testing remains an important diagnostic tool, but may need to be performed regularly following SSc diagnosis to detect changes suggesting ILD. Baseline screening with HRCT, as part of a wider clinical workup that includes lung function testing, is necessary to confirm an SSc-ILD diagnosis. We favour a definition of disease progression as a sustained change in lung function characterised by an FVC decline from baseline of ≥10%, or an FVC decline of 5–9% in association with a DLCO decline of ≥15%. This decline in FVC or DLCO should be considered as disease progression even if it is only observed beyond the 12-month period. Patient-reported outcome tools focused on breathlessness and imaging may provide valuable supporting information. In future, it is hoped that new imaging techniques and diagnostic and predictive biomarkers will play an increasingly important role in defining SSc-ILD progression.
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02026-2019.Shareable
Acknowledgements
Writing, editorial support and formatting assistance was provided by Tom Priddle and Claire Scofield of Nucleus Global, which was contracted and funded by Boehringer Ingelheim International GmbH. Boehringer Ingelheim International GmbH was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Footnotes
Author contributions: The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors. They take full responsibility for the scope, direction, content of, and editorial decisions relating to, the manuscript, were involved at all stages of development and have approved the submitted manuscript. The authors received no compensation related to the development of the manuscript. Boehringer Ingelheim International GmbH supported the meeting of the international panel of experts.
Conflict of interest: O. Distler reports personal fees for consultancy from Boehringer, during the conduct of the study; grants and personal fees for lectures from Actelion, personal fees for advisory board work from AbbVie, personal fees for consultancy from Acceleron Pharma, Anamar, Amgen, Catenion, CSL Behring, ChemomAb, Ergonex, GSK, Inventiva, Italfarmaco, iQvia, Medac, Lilly, Sanofi, Target BioScience, UCB, Baecon Discovery, Blade Therapeutics, Curzion Pharmaceuticals and Glenmark Pharmaceuticals, grants and personal fees for consultancy and lectures from Bayer and Boehringer Ingelheim, personal fees for lectures from iQone, Menarini, Mepha and Novartis, personal fees for consultancy and lectures from Medscape, MSD and Roche, grants and personal fees for consultancy from Mitsubishi, personal fees for consultancy and lectures and non-financial (travel) support from Pfizer, outside the submitted work; and has a patent US8247389, EP2331143 issued.
Conflict of interest: S. Assassi reports non-financial (writing) support from Boehringer Ingelheim, during the conduct of the study; grants from Biogen Idec and Bayer, grants and personal fees from Boehringer Ingelheim outside the submitted work.
Conflict of interest: V. Cottin reports personal fees for advisory board work and lectures and non-financial (travel) support from Actelion, grants, personal fees for advisory board work and lectures, and non-financial (travel) support from Boehringer Ingelheim and Roche, personal fees for advisory board work and data monitoring committee work from Bayer/MSD and Galapagos, personal fees for data monitoring committee work from Gilead, Celgene and Galecto, personal fees for advisory board work and lectures from Novartis, personal fees for lectures from Sanofi, personal fees for steering committee work and data monitoring committee work from Promedior, outside the submitted work.
Conflict of interest: M. Cutolo reports grants from Actelion, Boehringer Ingelheim, Horizon, BMS and Celgene, outside the submitted work.
Conflict of interest: S.K. Danoff reports grants and personal fees from Boehringer Ingelheim, grants from Genentech/Roche and Bristol Myers Squibb, personal fees from Galapagos and Galectic, during the conduct of the study.
Conflict of interest: C.P. Denton reports personal fees from Actelion, Bayer, Sanofi, Boehringer Ingelheim and Corbus, grants and personal fees from GlaxoSmithKline, Inventiva, CSL Behring and Leadiant Biosciences, during the conduct of the study.
Conflict of interest: J.H.W. Distler reports personal fees for speaking and advisory board work from Boehringer Ingelheim.
Conflict of interest: A-M. Hoffmann-Vold reports non-financial support from Boehringer Ingelheim, during the conduct of the study; grants, personal fees and non-financial support from Boehringer Ingelheim, personal fees and non-financial support from Actelion, personal fees from Roche, outside the submitted work.
Conflict of interest: S.R. Johnson reports grants from Boehringer Ingelheim, Corbus, Bayer, Roche and the Canadian Institutes of Health Research outside the submitted work.
Conflict of interest: U. Müller Ladner reports personal fees for advisory board work and lectures from Boehringer Ingelheim, outside the submitted work.
Conflict of interest: V. Smith reports speaker fees from Boehringer Ingelheim, during the conduct of the study; grants from Boehringer Ingelheim, research funding from Actelion Pharmaceuticals Ltd, Bayer AG, Hoffman-La Roche AG, Galapagos NV and Sanofi, outside the submitted work.
Conflict of interest: E.R. Volkmann reports personal fees from Boehringer Ingelheim, outside the submitted work.
Conflict of interest: T.M. Maher reports grants and personal fees from GSK and AstraZeneca, grants, personal fees and non-financial support from UCB, personal fees from Boehringer Ingelheim, Roche, Bayer, Prometic, Samumed, Galapagos, Celgene, Indalo, Pliant, Blade Therapeutics, Respivant, Novartis and Bristol-Myers Squibb, stock options from Apellis, outside the submitted work.
Support statement: S.R. Johnson has been awarded a Canadian Institutes of Health Research New Investigator Award; and is supported by the Oscar and Eleanor Markovitz Fund for Scleroderma Research of the Arthritis Research Foundation. V. Smith is a Senior Clinical Investigator of the Research Foundation – Flanders (Belgium) (FWO) (1.8.029.15N). The FWO was not involved in study design, collection, analysis and interpretation of data, writing of the report, nor in the decision to submit the article for publication. T.M. Maher is supported by a National Institute for Health Research Clinician Scientist Fellowship (NIHR ref: CS-2013-13-017) and is a British Lung Foundation Chair in Respiratory Research (C17-3). Writing, editorial support and formatting assistance was contracted and funded by Boehringer Ingelheim International GmbH. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received October 15, 2019.
- Accepted February 9, 2020.
- Copyright ©ERS 2020
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










