





Abstract
Obstructive pulmonary disease in patients with α1 antitrypsin (AAT) deficiency (AATD) occurs earlier in life compared with patients without AATD. To understand this further, the aim of this study was to investigate whether AATD presents with altered neutrophil characteristics, due to the specific lack of plasma AAT, compared with non-AATD COPD.
This study focussed on the neutrophil plasma membrane and, by use of label-free tandem mass spectrometry, the proteome of the neutrophil membrane was compared in forced expiratory volume in 1 s (FEV1)-matched AATD, non-AATD COPD and in AATD patients receiving weekly AAT augmentation therapy (n=6 patients per cohort). Altered protein expression in AATD was confirmed by Western blot, ELISA and fluorescence resonance energy transfer analysis.
The neutrophil membrane proteome in AATD differed significantly from that of COPD as demonstrated by increased abundance and activity of primary granule proteins including neutrophil elastase on the cell surface in AATD. The signalling mechanism underlying increased degranulation involved Rac2 activation, subsequently resulting in proteinase-activated receptor 2 activation by serine proteinases and enhanced reactive oxygen species production. In vitro and ex vivo, AAT reduced primary granule release and the described plasma membrane variance was resolved post-AAT augmentation therapy in vivo, the effects of which significantly altered the AATD neutrophil membrane proteome to that of a non-AATD COPD cell.
These results provide strong insight into the mechanism of neutrophil driven airways disease associated with AATD. Therapeutic AAT augmentation modified the membrane proteome to that of a typical COPD cell, with implications for clinical practice.
Abstract
Neutrophils from patients with COPD due to alpha-1 antitrypsin deficiency illustrate an altered membrane protein profile and primary granule exocytosis pattern compared to cells from COPD patients without AATD, a defect corrected by augmentation therapy http://bit.ly/3balrrT
Introduction
α1 Antitrypsin (AAT) is recognised as a potent inhibitor of serine proteinases, primarily neutrophil elastase [1]. AAT is produced mainly by hepatocytes and is released into the circulation, yielding a plasma concentration of approximately 1.5 g·L−1. AAT deficiency (AATD) gives rise to reduced levels of AAT and is a genetic risk factor for the development of chronic obstructive pulmonary disease (COPD). Emphysema, routinely characterised by computed tomography (CT) [2], is an important feature of AATD, classically panacinar in nature, in contrast to the mainly centrilobular disease seen in non-AATD COPD. AAT augmentation therapy, which involves intravenous infusion of plasma purified human AAT, demonstrated a reduced rate of lung density loss [3, 4]. Moreover, improved biochemical effects of double AAT dosing (120 mg·kg−1·week−1) in AATD was recently described [5].
Although lung disease in AATD patients typically starts at an earlier stage in life compared to patients with COPD without AATD, wide-ranging studies have been performed to discriminate between AATD and non-AATD COPD phenotypes. Such studies include evaluating complications and survival post-lung transplant [6], plasma and urine biomarkers [7, 8], pulmonary rehabilitation [2], quality of life [9] and differences in adjusting to illness [10]. Airway neutrophilia is a feature of COPD with and without AATD, with increased neutrophil numbers reported in epithelial lining fluid of non-smoking AATD individuals compared with healthy controls, and are implicated in many of the pathological features associated with the disease, including unopposed neutrophil elastolytic activity [11]. In addition, airway neutrophilia has been described in AATD subjects even with mild functional lung impairment [12]. A possible explanation for increased neutrophil responsiveness in AATD is put forward by studies demonstrating that AAT possesses key anti-inflammatory properties independent of anti-protease activity. This is particularly relevant in regards to modifying essential neutrophil functions including leukotriene (LT) B4-induced adhesion [13], chemotaxis in response to interleukin (IL)-8 [14], degranulation of secondary and tertiary granules via tumour necrosis factor (TNF)-α signalling [15], oxidative activation [16] and the ability of AAT to normalise proapoptotic signals in circulating neutrophils [17]. It has previously been shown that AAT binds neutrophil plasma membranes, localised to membrane lipid rafts [14]. This raised the question of whether AAT augmentation therapy in AATD–COPD patients could bind the circulating AATD cell in vivoand modify the AATD neutrophil to that of a non-AATD COPD type cell. For this analysis, we chose to examine the neutrophil plasma membrane, which represents the interface between the cell and its environment and in large part determines a cell's response to stimuli. The aim of this study was to perform the first proteomic analysis of plasma membranes of neutrophils from AATD–COPD compared with non-AATD COPD individuals, and to elucidate the impact of AAT augmentation therapy. Finally, our objective was to identify the consequence of altered membrane protein expression on neutrophil function.
Materials and methods
Study design
Ethical approval from Beaumont Hospital Institutional Review Board was acquired and written informed consent obtained from all study participants (approval number 18/52). Healthy control volunteers (table 1) showed no evidence of any disease and had no respiratory symptoms; none were taking medication, all non-smokers and all proven MM phenotype with serum AAT concentrations within the normal range (1.5 g·L-1). AATD patients (homozygous for the Z-allele, non-smokers) were recruited from the Irish Alpha-1 Antitrypsin Deficiency Registry (table 1) and were classified as healthy AATD individuals (forced expiratory volume in 1 s (FEV1) >80% predicted), AATD individuals with airway obstruction (FEV1<80% predicted with obstructive pattern on spirometry (i.e. FEV1/forced vital capacity (FVC) ratio <0.7) and emphysema on CT), or AATD patients on augmentation therapy receiving plasma purified AAT from CSL Behring (Zemaira) administered intravenously at a dosage of 60 mg·kg−1 body weight weekly. Non-smoking COPD patients (FEV1 <80% predicted, with obstructive pattern i.e. FEV1/FVC ratio <0.7 and emphysema on CT) with serum AAT concentrations within the normal range were recruited from Beaumont Hospital (table 1). In the 6 weeks prior to obtaining blood samples, all patients were exacerbation free.
Characteristics of healthy controls and patient groups
Neutrophil isolation and cell assays
The methods for blood sampling for plasma and neutrophil isolation, along with plasma membrane isolation, quantitative label-free liquid chromatography–tandem mass spectrometry LC-MS/MS, Western blot analyses and ELISA are outlined in the supplementary materials and methods. Fluorescence resonance energy transfer (FRET) analysis [18] and cytochrome c reduction assays [19] are described in detail in the supplementary materials and methods section.
Statistical analysis
Results are expressed as mean±sem of biological replicates or independent experiments as stated in each figure legend. Data analysis was performed using GraphPad PRISM 8.0 (San Diego, CA, USA). Student's paired t-test was used where distribution was normal and when comparisons were being made between two matched groups. One-way or two-way ANOVA was used for independent group comparisons, followed post hoc by Tukey's multiple comparison test where appropriate. Non-parametric Wilcoxon signed rank testing was employed where data were not normal (by Shapiro–Wilk test). Results were considered significant when p<0.05. Proteomics results were analysed using Progenesis software. One-way repeated measures (within-subject) ANOVA was used for dependent group comparisons. The p-values were adjusted using Benjamini–Hochberg false discovery rate (FDR) for every protein identified. Differential protein expression was defined as ≥1.5-fold change in expression with a p-value of <0.05.
Results
The proteome of the circulating neutrophil plasma membrane is altered in AATD compared with COPD
We sought to identify differences in the neutrophil plasma membrane proteome in people with AATD compared with non-AATD COPD (table 1). At the time of referral, all AATD patients had chest imaging that revealed emphysema and obstructive spirometry (mean FEV1 40.5±14.2% predicted) with a reduced diffusing capacity for carbon monoxide (mean DLCO 38.3±7.01% predicted). Circulating plasma AAT levels were determined by routine nephelometry, and were in the low range for all six AATD patients (0.29 g·L−1). Moreover, AAT phenotype testing by isoelectric focussing (IEF) revealed the characteristic Z-AAT protein as three IEF bands, Z2, Z4, Z6, with the classic cathodal shift (fig. 1a). FEV1-matched COPD individuals without potentially confounding comorbidities were also recruited (n=6). By IEF, two of these individuals were determined to be MZ heterozygotes and thus excluded from further analysis (n=4, mean FEV1 41±11.6% predicted). The neutrophil plasma membrane fraction was isolated by sucrose gradient ultracentrifugation with the purity of isolated fractions confirmed by Western blotting for CD16b (fig. 1b). Label free tandem MS/MS proteomic analysis was performed following digestion of proteins by trypsin. The average yield was 860 proteins (range 765 to 1013), with identified proteins ranging in size from 5 kDa to 434 kDa. Proteomic analysis of plasma membranes of COPD neutrophils compared to AATD demonstrated 15 proteins that were differentially expressed (>1.5-fold change; ANOVA p<0.05). Unsupervised Pearson hierarchical cluster analysis of differentially expressed proteins demonstrated increased abundance in plasma membrane fractions of AATD patients (fig. 1c). Notably, of the 15 differentially expressed proteins, eight were identified as constituents of neutrophil granules (supplementary table S1) [20]. Of particular interest, the neutrophil plasma membrane fraction from AATD individuals demonstrated an increased abundance of the key primary granule constituents myeloperoxidase (MPO, fold change 3.3; p=0.03) and bactericidal/permeability increasing protein (BPI, fold change 8.4; p=0.02) (fig. 1d). Collectively, these results indicate changes in the levels of membrane-associated proteins of circulating neutrophils from individuals with AATD with respect to COPD counterparts, with evidence of increased levels of proteins from primary granules.

The proteome of the circulating neutrophil plasma membrane is altered in α1 antitrypsin (AAT) deficiency (AATD) compared with normal chronic obstructive pulmonary disease (COPD). a) Isoelectric focussing patterns of AAT phenotypes. Healthy control MM AAT glycoforms (M2-M8) are denoted on the left. Glycoforms from an AATD patient homozygous for the Z-allele (Z2, Z4 and Z6) are shown on the right. Plasma levels of AAT (g·L−1) for the homozygous and heterozygous phenotype are indicated. b) Coomassie blue stained gel of healthy control neutrophils subjected to subcellular fractionation yielding a cytosol fraction (Cyt) and plasma membrane (PM) fraction. Western blotting employed antibody to CD16b as a marker of PM fraction purity. c) Hierarchical (Pearson) cluster analysis of DE proteins in the PM fraction of circulating neutrophils from AATD (forced expiratory volume in 1 s (FEV1) <80% predicted) (n=6) compared with non-AATD FEV1-matched COPD patients (n=4). Differential expression defined using normalised abundance, >1.5-fold difference and p<0.05 by ANOVA. Red indicates increased protein abundance, blue indicates decreased abundance. Myeloperoxidase (MPO) and bactericidal/permeability increasing protein (BPI) are highlighted. d) Abundance of MPO (left hand panels) and BPI in AATD PM fractions compared to COPD as determined by tandem mass spectrometry proteomic analysis (p<0.05, n=6 and n=4 subjects per group, respectively, ANOVA). Experiments illustrated in panels a and b are each representative gels.
Increased release of primary granule enzymes in AATD
Following the finding of increased levels of granule proteins associated with the plasma membrane of AATD neutrophils, the process of primary granule degranulation was compared between AATD and healthy control (HC) cells. Although MPO, a marker of primary granules, is equally expressed in whole cell lysates (supplementary figure S1), basal unstimulated and TNF/N-Formylmethionyl-leucyl-phenylalanine (fMLP) stimulated levels of released MPO and BPI, and the additional primary granule marker proteinase 3 (PR3) appeared increased in supernatants of AATD neutrophils (fig. 2a). As Western blotting is only semi-quantitative, ensuing experiments evaluated levels of MPO and neutrophil elastase release by ELISA. For this analysis, neutrophils of healthy controls, AATD (FEV1 >80%), AATD (FEV1 <80%) and non-AATD COPD patients matched for FEV1 were compared. Post-10 min stimulation and relative to unstimulated cells, results revealed significantly increased levels of degranulated extracellular MPO in cell supernatants of AATD (FEV1 <80%) compared with healthy controls (p=0.04) or AATD (FEV1 >80%) (p=0.03) (fig. 2b). Moreover, significantly increased levels of degranulated neutrophil elastase was detected in cell supernatants of AATD (FEV1 <80%) compared with healthy controls (p=0.0006) or non-AATD COPD (FEV1 <80%) (p=0.006) (fig. 2c). In addition, by investigating membrane-bound neutrophil elastase levels by FRET analysis, compared to healthy control cells, results revealed a significant increase in plasma membrane bound neutrophil elastase activity on AATD (FEV1 <80%) cells at rest (p=0.01) and after 10 min TNF/fMLP stimulation (p=0.01) (fig. 2d).
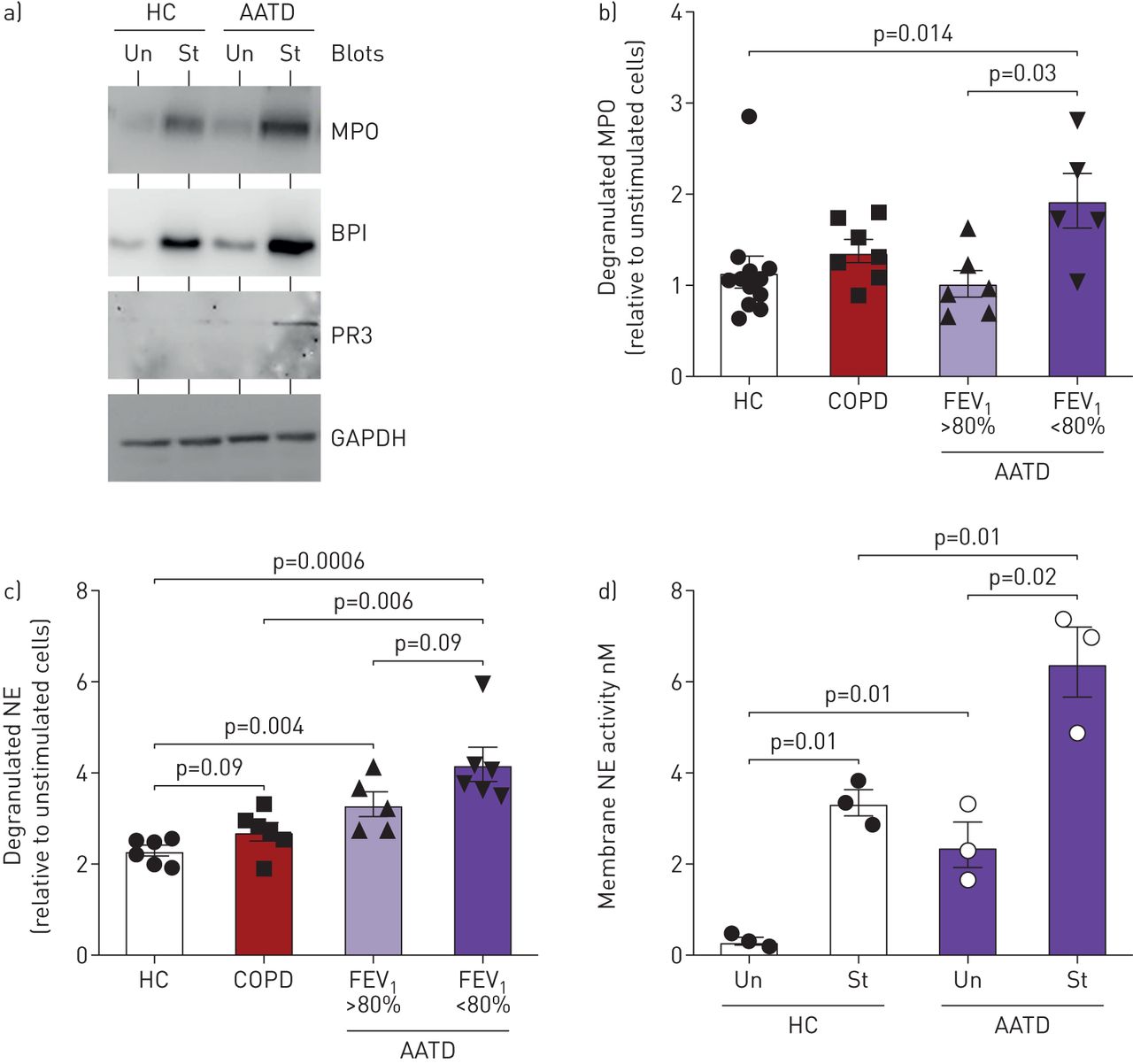
Increased primary granule release from α1 antitrypsin (AAT) deficiency (AATD) neutrophils. a) Neutrophils (1×107) isolated from healthy controls (HC) or AATD (forced expiratory volume in 1 s (FEV1) <80% predicted) individuals were incubated at 37°C and were either untreated (Un) or stimulated with tumour necrosis factor (TNF) (10 nM)/fMLP (100 nM) (St). Cell-free supernatants were collected and Western blotted for markers of primary granule release including myeloperoxidase (MPO), BPI and PR3. Representative blots of n=6 subjects per group. Western blot of unstimulated and stimulated neutrophil whole cell lysates for GAPDH demonstrated equal protein loading indicative of equal cell numbers used per reaction. b and c) a comparison of stimulated levels of MPO (b) and neutrophil elastase (c) release by healthy control (HC), AATD cells from individuals with FEV1 >80% predicted, AATD cells from patients with FEV1 <80% and matched non-AATD chronic obstructive pulmonary disease COPD patients, FEV1 <80% predicted. Significance was tested by one-way ANOVA, followed by Tukey's post hoc multiple comparison test. d) Increased neutrophil elastase activity was recorded on the outer plasma membrane of resting AATD (FEV1 <80%) neutrophils compared with resting HC cells (p=0.01 by unpaired t-test; n=3 subjects per group), with a significant increase observed in both groups in response to stimulation (St) with tumour necrosis factor (TNF)α/N-Formylmethionyl-leucyl-phenylalanine (fMLP) for 10 min (p=0.01, p=0.02, respectively). Measurements are mean±sem.
A central role of Rac2 in primary granule exocytosis has been described [21], with peak GTP-bound Rac2 activity occurring between 30 s and 5 min of stimulation [22]. In the current study, TNF/fMLP stimulated levels of active GTP-bound Rac2 were increased in neutrophil lysates of AATD (FEV1 <80%) individuals compared with healthy controls (p=0.02) (fig. 3a). Moreover, by inclusion of the Rac inhibitor, NSC23766 (50 μM) and quantification of extracellular MPO or neutrophil elastase by ELISA, MPO and neutrophil elastase degranulation by AATD cells was significantly reduced (p=0.01 and p=0.001, respectively) (fig. 3b). Collectively, these results demonstrate that Rac2 activation is altered in AATD neutrophils, which may contribute significantly to increased degranulation of primary granules.

Increased Rac2 activation in α1 antitrypsin (AAT) deficiency (AATD) neutrophils. a) Significantly increased expression levels of active GTP-bound Rac2 (upper panel) in tumour necrosis factor (TNF)/N-Formylmethionyl-leucyl-phenylalanine (fMLP) stimulated AATD (forced expiratory volume in 1 s (FEV1) <80%) neutrophils (1×107) compared with healthy control (HC) cells (p=0.02 by unpaired t-test; n=3 subjects per group). For quantification, the amount of Rac2-GTP is calculated relative to total Rac2 (bottom panel). b) AATD cells remained untreated or treated with the Rac inhibitor NSC23766 (50 µM) for 10 min and then stimulated with TNF/fMLP for 10 min. The level of myeloperoxidase (MPO) or neutrophil elastase degranulation was measured by ELISA and expressed as pM per 106 cells (n=12). Significance was tested by paired t-test or by Wilcoxon matched-pairs signed rank test.
Neutrophil elastase induced superoxide anion production
We sought to explore the consequence of increased primary granule exocytosis in AATD, with focus on neutrophil elastase mediated NADPH oxidase activation and enhanced O2− production. By use of a cytochrome c reduction assay, and when analysed with respect to the presence of airway obstruction, there was no difference in O2− production by unstimulated neutrophils of healthy control, healthy AATD individuals (FEV1 >80% predicted) or AATD with established airways disease (FEV1 <80% predicted) (fig. 4a). Moreover, fMLP (100 nM) only poorly activated the NADPH oxidase of healthy control or healthy AATD neutrophils, confirming the un-primed status of these cells (fig. 4a). In contrast, however, fMLP induced a significant 1.8-fold increase in O2− production by cells of AATD patients with established airways disease, when compared with unobstructed AATD cells (2.1±0.2 nmol O2− versus 1.2±0.3 nmol O2− per 106 cells, respectively; p=0.015) (fig. 4a). Moreover, when analysed with respect to TNF/fMLP stimulated activation, there was no difference observed in levels of O2− produced by healthy control compared with AATD cells from individuals with FEV1 >80% or non-AATD COPD patients (FEV1 <80%) (fig. 4b). In contrast, however, a significant difference was observed in O2− production by AATD cells from individuals with FEV1 <80% compared with healthy control cells (3.0±0.3 nmol O2− versus 2.1±0.2 nmol O2− per 106 cells, respectively; p=0.01) (fig. 4b).

neutrophil elastase cleavage of PAR2 triggers O2− production. A comparison of basal levels of O2−, measured by cytochrome c reduction, generated by healthy control (HC), α1 antitrypsin (AAT) deficiency (AATD) neutrophils from individuals with forced expiratory volume in 1 s (FEV1) >80% predicted, AATD cells from patients with FEV1 <80% and non-AATD chronic obstructive pulmonary disease (COPD) patients with FEV1 <80% predicted (106 cells per patient group). Cells remained untreated (Un) or treated with N-Formylmethionyl-leucyl-phenylalanine (fMLP) (100 nM) (a), or stimulated with tumour necrosis (TNF) (10 nM)/fMLP (100 nM) (b). AATD neutrophils (FEV1 <80%) produced significantly more O2−. Significance was tested using two-way ANOVA followed by Tukey's multiple comparison test. c) Flow cytometry analysis using a FITC-labelled anti-PAR2-antibody demonstrates significantly reduced levels of plasma membrane PAR2 post neutrophil elastase exposure (100 nM, 15 min) compared to untreated cells (p=0.01, Student's t-test, n=3). d) Cytochrome c assay of O2− production by HC neutrophils stimulated with neutrophil elastase (250 nM), neutrophil elastase plus AAT (5 or 27.5 µM) or neutrophil elastase following pre-treatment with PAR2 blocking Ab (Anti-PAR2; 10 µg·mL−1). Significance was tested using two-way ANOVA followed by Tukey's multiple comparison test. Bars are the mean±sem. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001
It has been shown that the presence of neutrophil elastase increases reactive oxygen species (ROS) production via cleavage of protease activated receptor 2 (PAR2) [23]. We hypothesised that the presence of neutrophil elastase on AATD neutrophils would result in increased production of ROS via cleavage of PAR2 from the cell surface. To confirm that neutrophil elastase could activate PAR2 on neutrophils, cells (1×106) were treated with neutrophil elastase (250 nM) and the level of PAR2 assessed by flow cytometry (supplementary figure S2). Neutrophil elastase treated neutrophils demonstrated a significant three-fold reduction in the presence of PAR2 on the cell surface (p=0.01) (fig. 4c), indicative of PAR2 activation [23]. To understand whether neutrophil elastase:PAR2 activation triggered ROS production, neutrophils of healthy control, non-AATD COPD patients (FEV1 <80%), AATD individuals with FEV1 >80% or AATD individuals with FEV1 <80% were challenged with TNF/fMLP, neutrophil elastase, neutrophil elastase in combination with AAT (5 or 27.5 µM) or a PAR2 blocking antibody (10 μg·mL−1) (fig. 4d). Healthy control neutrophils challenged with neutrophil elastase demonstrated a significant 2.1-fold increase in O2− production compared with untreated cells (p<0.0001), an effect inhibited by inclusion of 5 µM (p=0.05) or 27.5 µM AAT (p=0.0001) or the PAR2 blocking Ab (p<0.0001) (fig. 4d). Furthermore, neutrophils of AATD individuals with FEV1 <80% demonstrated significantly increased levels of O2− production in response to neutrophil elastase compared to healthy control cells (p=0.0005) or neutrophils of non-AATD COPD patients with matched FEV1 (p=0.03). The increased level of O2− production by cells of AATD individuals with airway obstruction was significantly inhibited by AAT (5 µM, p<0.0001; 27.5 µM, p<0.0001) and the PAR2 blocking Ab (p<0.0001) (fig. 4d). Collectively, these results corroborate neutrophil elastase mediated PAR2 activation and O2− production, highlighting a further dimension to the benefits of circulating plasma AAT.
The in vitro effect of exogenous AAT on dysregulated primary granule release by AATD neutrophils
We next investigated the effects of AAT in vitro on neutrophil degranulation. By immunoblot analysis of cell-free extracellular supernatants, levels of released MPO in response to TNF/fMLP stimulation were found to be significantly reduced in the presence of increasing concentrations of AAT. Over a concentration gradient of 2 µM to 27.5 µM, AAT exerted an inhibitory effect on TNF/fMLP mediated MPO release with a 50% decrease observed for 6.8 µM AAT, whilst 27.5 µM AAT further reduced MPO release by approximately 75% (p=0.02) (fig. 5a). The impact of exogenous AAT (2 or 27.5 μM) on TNF/fMLP induced primary granule degranulation of AATD cells was next examined. The increased degranulation of MPO after 5 min stimulation was significantly inhibited in healthy control and AATD cells by 27.5 μM AAT (p=0.02 and p=0.002, respectively) (fig. 5b). Collectively, these experiments demonstrate the ability of physiological levels of AAT to impact upon TNF/fMLP induced neutrophil degranulation processes, an immunomodulatory property compromised in AATD, and potentially impacted upon by AAT augmentation therapy, which was next explored.

Exogenous α1 antitrypsin (AAT) modifies neutrophil primary granule release in vitro. a) Neutrophils (1×107 cells·mL−1) isolated from healthy controls (HC) were stimulated with tumour necrosis factor (TNF) (10 nM)/N-Formylmethionyl-leucyl-phenylalanine (fMLP) (100 nM) in the absence or presence of increasing concentrations of AAT (2–27.5 µM). Cell-free supernatants were collected at 5 min and analysed by Western blotting for AAT or myeloperoxidase (MPO). Exogenous AAT reduced the degranulation of MPO in a concentration-dependent manner (p=0.02 by one-way ANOVA with Geisser–Greenhouse correction of densitometric units (DU)). Western blot of corresponding neutrophil whole-cell lysates for GAPDH demonstrated equal cell numbers used per reaction indicative of equal protein loading. b) Neutrophils of HC or AAT disease (AATD) patients were stimulated in the absence or presence of AAT (2 or 27.5 μM). Supernatants were analysed by Western blotting to determine the neutrophil degranulation pattern. AAT (27.5 μM) significantly reduced degranulation of MPO by HC and AATD cells (p=0.02 and p=0.002 respectively, Mann–Whitney U-test). All results are expressed as relative densitometry units. All measurements are mean±sem.
AAT augmentation therapy in vivo modifies the neutrophil plasma membrane proteome
Ensuing experiments investigated whether AAT augmentation therapy resulted in an altered neutrophil plasma membrane proteome in vivo, and specifically, whether results indicating modification of primary granule release by exogenous AAT in vitro, could be confirmed in vivo. Plasma and neutrophils were isolated from AATD patients on augmentation therapy (FEV1 42.9±13% predicted). 2 days post infusion, the circulating plasma levels of AAT were significantly increased, in comparison with levels at the nadir of prior treatment (∼1.4 g·L−1 and ∼0.8 g·L−1, respectively; p<0.0001) (fig. 6a). To understand whether infused AAT binds to circulating neutrophil plasma membrane, the membrane AAT expression profile of AATD neutrophils was assessed by flow cytometry (fig. 6b). Results revealed a significant increase in the level of membrane bound AAT detected on day 2 post therapy (mean fluorescence=39) compared with day 0 pre-therapy (mean fluorescence=10.2; p<0.001). We performed proteomic analysis of the plasma membrane fraction of neutrophils isolated from the circulation of AATD individuals receiving AAT augmentation therapy at their nadir AAT levels (day 0) and paired samples on day 2 following treatment (n=6). Tandem MS/MS proteomic analysis identified 66 intrinsic neutrophil proteins that were differentially expressed between day 0 and day 2 of AAT augmentation therapy (fold change >1.5; within subject ANOVA p≤0.05). Of these, 65 were increased in abundance on day 0. Hierarchical cluster analysis was performed on differentially expressed proteins and presented by heat map (fig. 6c). This visual representation demonstrates the differences seen in the neutrophil plasma membrane proteome at these time-points of AAT augmentation therapy.

α1 Antitrypsin (AAT) augmentation therapy modifies the neutrophil proteome in vivo. a) Increased plasma levels of AAT in AAT disease (AATD) patients 2 days (D2) post receiving AAT augmentation therapy compared to levels at the nadir of prior treatment (D0) (p<0.0001, n=11 and n=6, respectively, Students' t-test). b) Cells were incubated with FITC-labelled goat polyclonal anti-AAT (1 μg/106 cells) and fluorescence determined by flow cytometry. Representative image of three independent experiments demonstrating increased levels of membrane-bound AAT on isolated AATD neutrophils 2 days post augmentation therapy (D2, red) compared with day 0 (D0, green). Isotype control presented in blue. c) Hierarchical cluster analysis of differentially expressed proteins from proteomic analysis of the PM fraction of circulating neutrophils from AATD individuals on AAT augmentation therapy (n=6, AATD #1–6) on D0 pre-treatment and D2 post-treatment. Heat map demonstrating differentially expressed proteins. Differential expression defined using normalised abundance, >1.5-fold difference and p≤0.05 by within-subject ANOVA. Red indicates increased protein abundance, blue indicates decreased abundance. Myeloperoxidase (MPO) and PR3 are highlighted. d) Tandem mass spectrometry proteomic analysis of neutrophil PM fractions of AATD patients isolated on D2 post-receiving augmentation therapy demonstrated a significant decrease in levels of MPO and PR3 compared with neutrophils isolated on D0 (p=0.05 and p=0.005 respectively, n=6 subjects, within-subject ANOVA).
Gene ontology biological process cluster analysis was performed on differentially expressed proteins using the online gene ontology tool GORILLA [24]. Gene ontology cluster analysis commonly results in redundant terms and thus the online REVIGO (Reduce and Visualise Gene Ontology) tool was used to summarise and visualise results [25]. REVIGO was used to generate a network graph of enriched biological process clusters (supplementary figure S3). Of most significance to the current study, the differentially expressed group was enriched for proteins involved in exocytosis (gene ontology term 0006887, enrichment score 1.64; p=0.0008, FDR adjusted q=0.03). 31 of the 65 differentially expressed intrinsic neutrophil proteins were associated with this gene ontology term (supplementary table S2). Individual proteins are represented by their UniProtKB number, with MPO and proteinase-3 (PR3) highlighted as representative primary granule proteins. Most notably, the archetypal constituents of primary granules MPO and PR3 were increased on day 0 compared to day 2 of AAT augmentation therapy (p≤0.05, fold change 2.6 and 2.7 respectively) (fig. 6d).
We next aimed to validate proteomic results by Western blot. Neutrophil membranes were isolated on day 0 and day 2 of AAT augmentation therapy (n=6 per group). Immunoblot densitometry demonstrated significantly decreased abundance of MPO (p=0.02) and PR3 (p=0.02) in membrane fractions of AATD patients on day 2 compared with day 0 of AAT augmentation therapy (fig. 7a). In concurrence, results of FRET analysis revealed that AATD neutrophils illustrated significantly reduced levels of membrane associated neutrophil elastase activity on day 2 post treatment when compared with neutrophils of patients not on therapy (p=0.007) (fig. 7b). In addition, there was a significant decrease in levels of active GTP-bound Rac2 on day 2 post-augmentation therapy when compared with day 0 pre-treatment (p=0.04) (fig. 7c). Moreover, upon TNF/fMLP activation of purified neutrophils from AATD individuals on day 0 and day 2 of AAT augmentation therapy, significantly reduced levels of MPO were degranulated to the extracellular milieu on day 2 of AAT augmentation therapy (p=0.007) (fig. 7d). Of note, the level recorded in the augmentation therapy group was not statistically different when compared to the level released by healthy control cells, suggesting normalisation of the degranulation process post-therapy (fig. 7d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
α1 Antitrypsin (AAT) augmentation therapy alters the neutrophil membrane proteome of AAT disease (AATD) cells to that of chronic obstructive pulmonary disease (COPD) neutrophils. a) Western blot demonstrating decreased abundance of myeloperoxidase (MPO) (n=6) and PR3 (n=4) in neutrophil PM fractions on day 2 (D2) of AAT augmentation compared with day 0 (D0) (p=0.02). Na+/K+ ATPase was found equally expressed between the different membrane types and was therefore used as a loading control. b) Decreased neutrophil elastase activity was observed on neutrophil plasma membranes post AAT augmentation therapy (+aug) compared to no treatment (-aug) (p=0.007 by unpaired t-test; n=4 subjects per group). c) Significantly decreased expression levels of active GTP-bound Rac2 (upper panel) in AATD neutrophils on D2 post augmentation therapy compared to pre-treatment D0 (p=0.04 by paired t-test, n=3 subjects per group). Active Rac2-GTP is calculated relative to total Rac2 (bottom panel). d) Neutrophils (1×107) isolated from healthy control (HC) or AATD individuals on D0 or D2 of AAT augmentation therapy were stimulated with tumour necrosis factor (TNF) (10 nM)/N-Formylmethionyl-leucyl-phenylalanine (fMLP) (100 nM). AATD neutrophils released significantly less MPO on D2 compared with D0 (p=0.007 by paired t-test of densitometric units (DU); n=3 subjects per group). Western blot of unstimulated and stimulated neutrophil whole cell lysates for GAPDH demonstrated equal protein loading indicative of equal cell numbers used per reaction (bottom panel). e) Heatmap demonstrating differentially expressed proteins in the plasma membrane fraction of circulating neutrophils from COPD patients compared with AATD individuals (n=6) on D0 (pre-treatment) and D2 of augmentation therapy. Differential expression defined using normalised abundance, >1.5-fold difference and p<0.05 by ANOVA. 14 proteins were increased in expression in AATD D0 compared to AAT sufficient COPD individuals, with one protein remaining differentially expressed on D2 (EH-domain containing protein 1, fold change 2.9, p=0.03). Each measurement is the mean±sem from biological replicates.
To investigate whether AAT augmentation therapy converted the AATD neutrophil membrane proteome to that of an AAT sufficient COPD cell, thereby confirming the effect of AAT augmentation in vivo, we next compared the plasma membrane proteome of neutrophils isolated from COPD to that of AATD individuals (n=6) 2 days post-intravenous augmentation therapy (day 2) (fig. 7e). Only one protein was significantly differentially expressed in this analysis (EH domain-containing protein 1, fold change 2.9; p=0.03), suggesting that AAT augmentation therapy caused reversion of the PM proteome to a state more analogous to that of the AAT sufficient COPD neutrophil (fig. 7e). Furthermore, granule constituent proteins were not differentially expressed in this comparison. Collectively, these results illustrate the effect of weekly AAT infusions on Rac2 activation and degranulation in vivo, both of which are clearly related to the levels of AAT in plasma.
Discussion
This study demonstrates the ability of AAT augmentation therapy to impact on the membrane proteome of AATD–COPD neutrophils, and elucidates the mechanism by which its deficiency leads to increased primary granule release, resulting in PAR2 activation and ROS generation.
By use of tandem MS analysis, we demonstrated that the membrane proteome of circulating neutrophils differs between AATD–COPD and FEV1-matched non-AATD COPD individuals. Proteomic results indicated increased primary granule proteins including MPO and BPI, interacting with the neutrophil plasma membrane. Accordingly, we show that at baseline and also in response to stimulation, neutrophil primary granule degranulation capacity was significantly higher in AATD patient neutrophils, with elevated levels of MPO, BPI, PR3 and neutrophil elastase released compared with control cells. In line with this result, significantly increased levels of MPO and neutrophil elastase were detected in AATD–COPD patient sputum compared with AAT-sufficient COPD individuals [26]. The clinical implications of increased MPO release have been demonstrated by the use of an MPO inhibitor, which reduced progression of emphysema and small airway remodelling while partially protecting against pulmonary hypertension, suggesting a potential therapeutic role for MPO inhibitors in the management of obstructive airways disease [27]. The current study also demonstrated increased neutrophil elastase activity on the surface of AATD–COPD neutrophils compared with healthy controls. Dysregulated neutrophil elastase activity has previously been shown to induce secretion of proinflammatory stimuli including LTB4 and IL-8 [11], thus generating a cycle of inflammation.
It has previously been documented that neutrophils from patients with cystic fibrosis (CF) release greater levels of neutrophil elastase compared with healthy control cells [28]. Results of the present study further expands this field, and demonstrates that this phenomenon is not specific to CF, and that AATD–COPD cells also release significant levels of primary granules, with increased levels of MPO, BPI and neutrophil elastase detected on membranes of AATD cells, underscoring the assertion that inflammation rather than intrinsic cellular defects may be the cause. Degranulation of MPO and neutrophil elastase by neutrophils from obstructed AATD individuals was elevated, to a proportionately greater extent than healthy control cells, congruent with elevated levels of circulating pro-inflammatory mediators recorded in AATD [5]. It is recognised that granule proteins remain bound to the cell surface of neutrophils when granules fuse with the plasma membrane during exocytosis. Indeed, it has been shown that MPO binds membranes via a CD11b/CD18 integrin-dependent mechanism [29], and neutrophil elastase can rebind membranes and exosomes derived from neutrophils [30]. Of note however, although neutrophil elastase was found associated with neutrophil plasma membranes by FRET analysis, proteomic examination did not identify a difference in neutrophil elastase. This anomaly may possibly arise due to neutrophil elastase autolysis during protein sample preparation for mass spectrometry. Neutrophil degranulation of primary granules is regulated by Rac2 and in the present study measurements of GTP-bound Rac2 illustrated significantly increased activity in AATD neutrophils. Interestingly, although both CF [28, 31] and AATD–COPD neutrophils secrete increased levels of primary granules, no such similarity exists for secondary and tertiary granule release; compared with healthy control cells, AATD [15] and CF cells [32] release significantly more and less, respectively. In CF, it was demonstrated that decreased Rab27a activation lead to decreased secondary and tertiary granule release, highlighting the different roles that Ras proteins play in the regulation of neutrophil degranulation and the need for further studies exploring their regulation and involvement in airways disease.
The observed excessive neutrophil degranulation by AATD–COPD cells prompted us to question the consequence and possible impact on disease progression. This is not a straightforward concept, however, as not all AATD patients suffer from emphysema or COPD and even in those with severe AAT deficiency, the development of lung disease is variable and most likely involves environmental, occupational or genetic modifiers. Nonetheless, airway oxidative stress has been associated with air trapping, bronchitic symptoms and loss of lung function [33]. Based on previous findings of ROS generation following neutrophil elastase activation of PAR2 on nociceptive neurons [23], we explored neutrophil elastase activation of PAR2 on neutrophils with results demonstrating significant superoxide anion production. The described signalling axis was inhibited by inclusion of a PAR2 blocking antibody and implies that PAR2 antagonists may be used as a novel anti-oxidant therapy in obstructive airways disease where PAR2 plays a role [34]. Alternatively, a second approach could involve modulation of neutrophil primary granule release, thereby removing the source of the PAR2 stimulant. Consistent with the results indicating an exaggerated degranulation response by AATD–COPD neutrophils in the current study, physiological levels of AAT significantly reduced the level of primary granule exocytosis in vitro, as demonstrated by reduced detectable levels of released MPO. The effect of AAT on neutrophil O2− production was expanded to a midpoint of 5 μM. However, whilst this concentration of AAT inhibited superoxide production in vitro, this may not be the case in vivo, as mutant Z-AAT of AATD patients may polymerise, and possess reduced anti-elastase capacity [35]. The benefits of the immune-regulatory capacity of AAT in modulating primary granule release may extend beyond prevention of PAR2 activation, as AATD is linked to development of autoimmunity and anti-neutrophil cytoplasmic antibodies (ANCA) [36, 37]. The main antigenic targets for ANCA include components of neutrophil primary granules including exocytosed MPO, PR3 [38] and more infrequently, neutrophil elastase [39].
To support in vitro results demonstrating the ability of AAT to modulate primary granule release, we isolated plasma and neutrophils from AATD–COPD patients who were receiving AAT augmentation therapy. Results revealed that 2 days post-augmentation therapy levels of circulating AAT were within the healthy control normal range (27.5 μM), and significantly increased levels of infused AAT were detected bound to membranes of the circulating AATD neutrophil. Neutrophil plasma membrane proteome and gene ontology cluster analysis of differences between day 0 and day 2 of AAT augmentation therapy revealed decreased plasma membrane expression of MPO and PR3, indicative of reduced primary granule release. Proteomic results were confirmed by Western blotting, with AAT therapy shown to reduce exuberant Rac2 activation, as shown by the decreased level of bound GTP. In our study, AAT treatment equalised the AATD-COPD neutrophil plasma membrane proteome to that of COPD cells, as indicated by aligned expression of all but one protein initially shown differentially expressed in the day 0 comparison. The conclusion of the results is clear, and demonstrate that the neutrophil membrane in AATD patients compared with non-AATD deficient COPD patients with matched FEV1 is altered, most probably due to a lack of circulating AAT. One further point to emphasise is that the effects of AAT were most prominent at day 2 of therapy and almost all worn off by day 0. Moreover, findings from the RAPID (Randomized, Placebo-controlled Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency) clinical trial confirmed the benefits of 60 mg·kg−1·week−1 AAT therapy in slowing progression of emphysema radiologically, however augmentation therapy does not halt the decline in lung function. This suggests the possible need to re-evaluate the set dose of 60 mg·kg−1, and that increased AAT dosing than currently exercised may lead to enhanced clinical benefits, as recently suggested [5].
A number of limitations to this study should be discussed. Firstly, the significance of mid-level AAT plasma levels and mutated forms of the human AAT protein, as observed in MZ- and SZ-AATD phenotypes, on neutrophil primary granule degranulation has not been fully addressed, though our in vitro data suggest a titratable phenotype that improves with increasing AAT. The prevalence of the MZ and SZ phenotypes of AATD are considerably greater than the ZZ phenotype assessed in this study. Thus, additional research is needed to clarify alterations in the neutrophil plasma membrane, considering the high number of individuals who are heterozygous for AATD [40]. In addition, due to the recruitment criteria a second limitation of this study is the participation of a relatively small number of AATD patients receiving augmentation therapy. Despite this drawback, however, this study provides evidence of the impact of infused AAT on the plasma membrane proteome and degranulation process of the circulating neutrophil.
In summary, neutrophils from people with AATD with the most severe ZZ mutation illustrate an altered membrane protein profile caused by increased Rac2 activation, imparting excessive primary granule degranulation. This defect was corrected by treatment with AAT augmentation therapy, the effects of which significantly altered the membrane proteome to that of a non-AATD COPD cell. These results highlight the important role that AAT plays in regulating neutrophil biology and the importance of AAT augmentation therapy in management of patients with AATD.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary methods and materials erj-01678-2019.supplement
Supplementary figures erj-01678-2019.figures
Supplementary table S1 erj-01678-2019.table_S1
Supplementary table S2 erj-01678-2019.table_S2
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01678-2019.Shareable
Acknowledgements
We thank the patients and healthy volunteers who graciously participated in this study.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: S. Landers, T. McEnery, K. McQuillan, M.P. Murphy, O. Coleman, P. Hawkins, M. Henry, P. Meleady, E.P. Reeves and N.G. McElvaney contributed to study design. S. Landers, T. McEnery, K. McQuillan, M.P. Murphy, O.J. McElvaney, O.F. McElvaney, A. Bussayajirapong, O. Coleman and M. Henry performed experiments, analysed and interpreted the data. T. McEnery, E.P. Reeves and N.G. McElvaney wrote the manuscript.
Conflict of interest: T. McEnery has nothing to disclose.
Conflict of interest: K. McQuillan has nothing to disclose.
Conflict of interest: O.F. McElvaney has nothing to disclose.
Conflict of interest: O.J. McElvaney has nothing to disclose.
Conflict of interest: S. Landers has nothing to disclose.
Conflict of interest: O. Coleman has nothing to disclose.
Conflict of interest: A. Bussayajirapong has nothing to disclose.
Conflict of interest: P. Hawkins has nothing to disclose.
Conflict of interest: M. Henry has nothing to disclose.
Conflict of interest: P. Meleady has nothing to disclose.
Conflict of interest: E.P. Reeves reports grants from Us Alpha-1 Foundation, during the conduct of the study.
Conflict of interest: N.G. McElvaney reports grants from MRCG HRB Funding scheme, during the conduct of the study.
Conflict of interest: M.P. Murphy has nothing to disclose.
Support statement: In support of this work, E.P. Reeves acknowledges funding from the US Alpha One Foundation and the Medical Research Charities Group/Health Research Board Ireland. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received August 23, 2019.
- Accepted January 30, 2020.
- Copyright ©ERS 2020
References










