





Abstract
Phosphatidylinositol 3-kinases (PI3Ks) and mammalian target of rapamycin (mTOR) play a role in the pathogenesis of idiopathic pulmonary fibrosis (IPF). Omipalisib (GSK2126458) is a potent inhibitor of PI3K/mTOR.
A randomised, placebo-controlled, double-blind, repeat dose escalation, experimental medicine study of omipalisib in subjects with IPF was conducted (NCT01725139) to test safety, tolerability, pharmacokinetics and pharmacodynamics. Omipalisib was dosed at 0.25 mg, 1 mg and 2 mg twice daily for 8 days in four cohorts of four subjects randomised 3:1 to receive omipalisib or placebo (two cohorts received 2 mg twice daily).
17 subjects with IPF were enrolled. The most common adverse event was diarrhoea, which was reported by four participants. Dose-related increases in insulin and glucose were observed. Pharmacokinetic analysis demonstrated that exposure in the blood predicts lung exposure. Exposure-dependent inhibition of phosphatidylinositol 3,4,5 trisphosphate and pAKT confirmed target engagement in blood and lungs. 18F-2-fluoro-2-deoxy-d-glucose(FDG)-positron emission tomography/computed tomography scans revealed an exposure-dependent reduction in 18F-FDG uptake in fibrotic areas of the lung, as measured by target-to-background, ratio thus confirming pharmacodynamic activity.
This experimental medicine study demonstrates acceptable tolerability of omipalisib in subjects with IPF at exposures for which target engagement was confirmed both systemically and in the lungs.
Abstract
Omipalisib, a PI3K/mTOR inhibitor, engages the target systemically and in the lungs of subjects with IPF. These effects were demonstrated in an experimental medicine study and were measured by inhibition of pAKT/AKT, PIP3/PIP2 and 18F-FDG-PET. http://ow.ly/7dMu30mZvcS
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fatal interstitial lung disease of unknown aetiology [1]. Two approved medicines that slow the rate of decline in pulmonary function (pirfenidone and nintedanib) are available [2, 3]. However, apart from lung transplantation, there is currently no treatment that halts or reverses decline in pulmonary function.
IPF and lung cancer share several cellular signalling pathways and transcriptional and epigenetic signatures [4, 5]. 18F-fluoro-2-deoxy-d-glucose (FDG)-positron emission tomography (PET) is used in oncology as a diagnostic, prognostic and theranostic endpoint [6, 7]. Increased uptake of 18F-FDG has been observed in areas of active fibrosis in IPF [8, 9]. In addition, increased lactate has been measured in the lungs of subjects with IPF, providing evidence of increased glycolytic metabolism [8, 10, 11]. Together, these observations support the rationale for exploring glucose metabolism in IPF as a therapeutic target and for using 18F-FDG-PET as a pharmacodynamic marker.
The class 1 phosphatidylinositol 3-kinases (PI3Ks) lipid kinases form a key oncogenic signalling node that is critical for glucose metabolism, cell growth, proliferation and survival [12, 13]. Omipalisib, a potent and selective inhibitor of the PI3K/mammalian target of rapamycin (mTOR) pathway [14], has been evaluated in a phase I clinical trial in subjects with solid tumours or lymphoma (www.clinicaltrials.gov NCT00972686). This signalling axis is being pursued as a target for anti-tumour therapy [14, 15]. A role for this pathway in IPF has been identified [11]. Detailed in vitro pharmacology demonstrates that omipalisib attenuates fibroblast proliferation and transforming growth factor (TGF)-β induced collagen synthesis in primary human lung fibroblasts and ex vivo precision-cut lung slices derived from IPF lung tissue [11]. These data were integrated with human pharmacokinetic data from oncology trials to inform a pharmacokinetic/pharmacodynamic model and predict a dosing regimen for effective PI3K engagement in the lungs of IPF subjects. In addition, cells derived from bronchoalveolar lavage (BAL) of subjects with IPF were tested for their potential as biosensors of pulmonary target engagement [11].
Having established a rationale for investigating the inhibition of the PI3k/mTOR pathway in IPF, we now describe the pharmacokinetic, pharmacodynamic and safety data from an experimental medicine study investigating the effects of short-term dosing of omipalisib in subjects with IPF.
Materials and methods
Study design
This was a double-blind, placebo-controlled, dose-escalation study of omipalisib in subjects with IPF. The study was designed to explore the pharmacology of a range of short-term doses of omipalisib. Signed informed consent was obtained from each subject prior to the performance of any study-specific procedures. Each cohort consisted of four subjects who were randomised 3:1 to receive omipalisib or placebo for 7–10 days. Subject and investigator were blinded to study treatment. Pharmacokinetics and pharmacodynamics were measured for up to 8 h post-dose. Safety and tolerability were assessed throughout the study. First and last daily doses were administered in the clinic and other doses were taken by subjects at home. Participants received hand-held spirometers and instructions on action to be taken in case of a >10% drop in forced vital capacity (FVC) or the development of new or worsening symptoms.
18F-FDG-PET/computed tomography (CT) scans and BAL were conducted twice during the study: once ≥2 days before dosing commenced and again between days 4 and 8 of dosing. PET/CT scan was always conducted before the BAL procedure to minimise interference of the BAL procedure on PET signals.
As previously described [16], BAL involved instillation of four successive 60 mL aliquots of 0.9% saline into a subsegment of the right middle lobe. BAL fluid (BALF) was manually aspirated, pooled, mixed and transferred to the laboratory on ice.
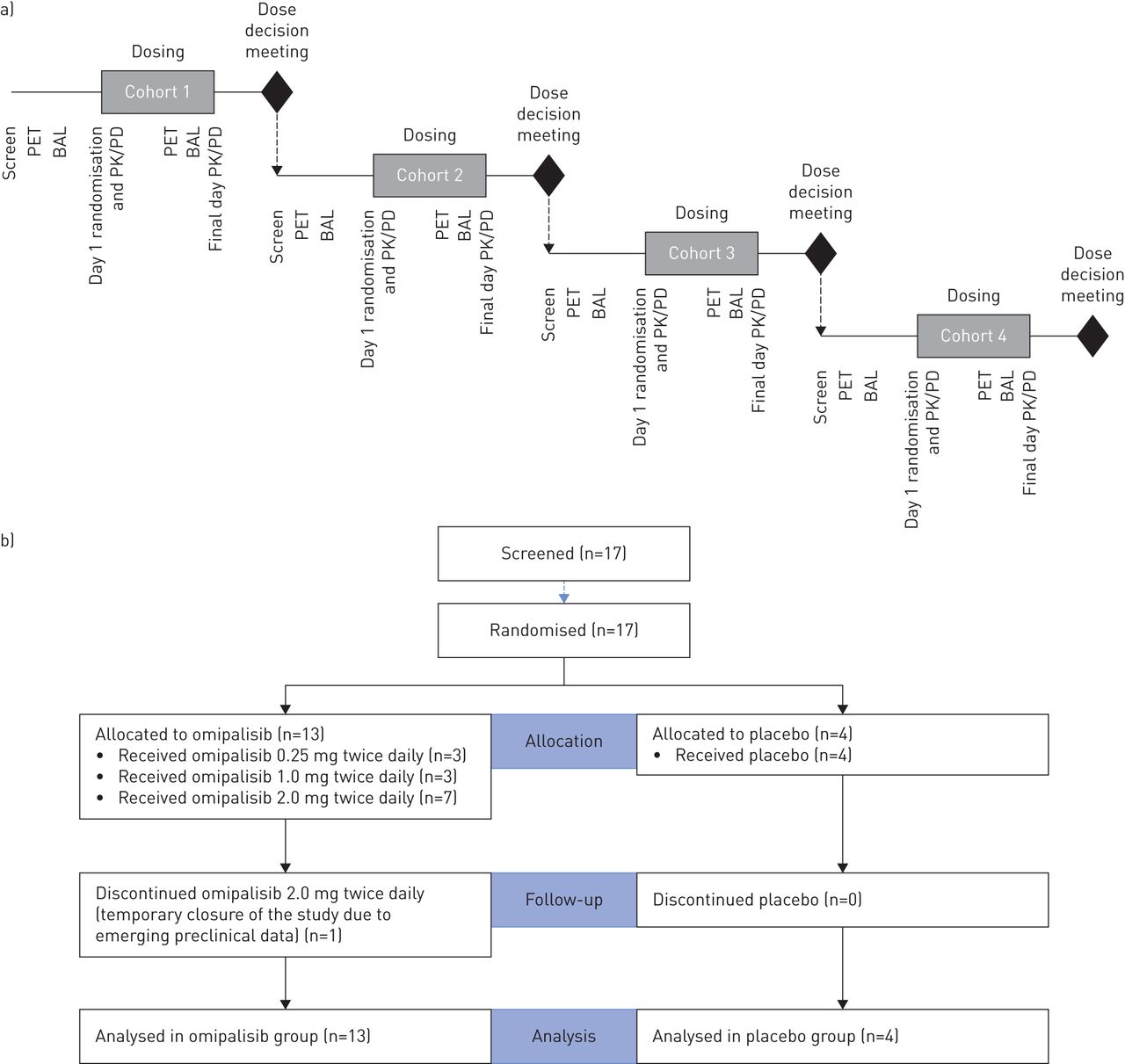
After the final subject in each cohort had completed dosing, a dose-escalation meeting was conducted, and safety, tolerability and pharmacokinetic data were reviewed by a safety committee (consisting of principal investigator, medical monitor, pharmacokineticist and statistician). The study statistician and pharmacokineticist were unblinded at the end of each cohort to conduct the analyses required for dose-escalation decisions. A schematic representation of the study design is provided (figure 1).

Schematic of the study design and CONSORT (Consolidated Standards of Reporting Trials) diagram. a) Each cohort consisted of four subjects randomised 3:1 to active agent and placebo. Doses of 0.25 mg twice daily or placebo were administered in cohort 1, doses of 1.0 mg twice daily or placebo were administered in cohort 2 and doses of 2.0 mg twice daily or placebo were administered in cohorts 3 and 4; b) CONSORT diagram. PET: positron emission tomography; BAL: bronchoalveolar lavage; PK: pharmacokinetics; PD: pharmacodynamics
The primary objective was to define the blood and pulmonary pharmacokinetic/pharmacodynamic relationship for omipalisib in IPF subjects. Endpoints measured included pAKT/AKT in platelet-rich plasma (PRP), pAKT/AKT and phosphatidylinositol 3,4,5 trisphosphate (PIP3)/phosphatidylinositol 4,5 bisphosphate (PIP2) in BAL cells, mean and 99th percentile standardised uptake value (SUVmean and SUV99%, respectively) of 18F-FDG-PET/CT, omipalisib pharmacokinetic parameters in blood (area under concentration–time curve (AUC), maximum observed concentration (Cmax), predose (trough) concentration at the end of the dosing interval (Ctrough)) and omipalisib concentration in BAL fluid and cells. Secondary objectives included safety, tolerability and cough (Leicester Cough Questionnaire [17, 18]). Given the short duration of the study, no formal assessment of antifibrotic efficacy was made.
Regulatory and ethics approval
The study was approved by the West London & GTAC research ethics committee (12/LO/1634) and the Medicines & Healthcare Products Regulatory Agency (MHRA) (European Union Drug Regulating Authorities Clinical Trials identification number 2012-001376-11). Permission to administer radioisotopes was obtained from the UK Administration of Radioactive Substances Advisory Committee (reference 630/3925/30809). The study was conducted at a single centre (Royal Brompton Hospital, London, UK) with PET/CT scans performed at University College London Hospital, Institute of Nuclear Medicine.
Study participants
Participants with a multidisciplinary team diagnosis of IPF according to international consensus criteria [1] and who met the following key inclusion criteria were eligible. FVC >40% predicted and diffusing capacity of the lung for carbon monoxide >30% pred; left ventricular ejection fraction ≥50%; and males aged >45 years and females >50 years (this difference reflecting varying risks of radiation exposure between sexes) with a body weight of ≥50 kg and ≥40 kg, respectively, and body mass index between 20 and 35 kg·m−2. The main exclusion criteria included unstable cardiovascular disease, IPF exacerbation, poorly controlled diabetes and current or previous use (within 30 days) of pirfenidone (nintedanib was not approved at the time the study was conducted).
Study drug
Omipalisib tablets were administered orally twice daily. A dose range of between 0.25 mg twice daily and up to 2 mg twice daily was predicted to provide pharmacologically relevant exposures as modelled using in vitro pharmacology and existing human pharmacokinetic data derived from oncology studies [11].
Pharmacokinetics
Full pharmacokinetic profiles were conducted on day 1 and on the last day of dosing. In addition, single mid-study pharmacokinetic samples were taken before the BAL and PET procedures. Blood and BAL samples were analysed for omipalisib using a validated analytical method based on protein precipitation, followed by high performance liquid chromatography/mass spectrometry (MS)/MS analysis.
A population pharmacokinetic modelling approach was utilised to characterise the blood pharmacokinetic and the total systemic exposure of omipalisib at each dose. A previously described pharmacokinetic model [11] was used as the base model and evaluated and updated using the pharmacokinetic data collected in this study. Cmax, Ctrough and 24-h AUC (AUC0-24) following each dose were predicted from the model, and were summarised. BAL cell pellet concentrations were adjusted by total cell counts in the BAL samples (mean±sd 0.26±0.20 million cells·mL−1 BALF).
Pharmacodynamic endpoints
Intracellular inhibition of PI3K in BAL cells and PRP was determined by measuring inhibition of AKT phosphorylation, assessed using meso scale discovery assay, Phospho (Ser473)/Total AKT Whole Cell Lysate Kit (Meso Scale Diagnostics, Rockville, MD, USA) and expressed as a ratio of the total AKT signal within each well with target engagement by omipalisib being expected to decrease phosphorylation of AKT. Platelets can be used for measuring pAKT as a surrogate for PI3K activity in blood [19].
PIP3 in snap-frozen BAL cells was assessed as PIP3 peak area by mass spectrometry at the Babraham Institute [20] and expressed as a ratio to PIP2 peak area. Inhibition of Pi3K by omipalisib decreases phosphorylation of PIP2 to PIP3, thereby increasing the ratio of PIP2 to PIP3.
Subjects underwent an 18F-FDG-PET/CT scan as described [21, 22]. Image analysis of whole lungs was the planned analysis for the 18F-FDG-PET/CT data. Image reconstruction and analysis was performed as previously outlined, using combined CT images for both attenuation correction and computation of the air fraction, to derive the SUVmean, SUV99% and Patlak-determined influx rate constant (Ki) [21–25]. SUV99% (the 99th percentile of SUV values in the lung mask) represents the maximum value in the lung while avoiding outlier pixels.
A post hoc target-to-background ratio (TBR) analysis was conducted, after unblinding of the study but without any reference to the treatment received, using methods described previously [26]. For each participant, the 3-cm3 diseased region with the highest uptake and a 3-cm3 region of normal tissue with minimal uptake in the same lung were identified on baseline static PET and confirmed on CT. These regions were automatically propagated to registered PET images of the mid-study scan. The TBR was calculated as the ratio of SUVmean in the disease region relative to SUVmean in the normal region, both with and without air fraction correction. Effective inhibition of Pi3K would be expected to decrease glucose uptake in metabolically active cells and thus decrease FDG-PET signal.
Sample size estimation
A simulation exercise was used to assess the probability of identifying a significant (at the p=0.05 level) concentration–response relationship between omipalisib and pAKT/AKT in BAL. In vitro data from BAL cells from six IPF donors from the PROFILE study [27] were used to simulate a concentration–response relationship when utilising three active participants at each chosen dose level. Based on this exercise, >99% of the simulated studies returned a significant concentration–response relationship, supporting the assumption that the chosen sample size would be appropriate.
Statistical methods
The primary aim of the study was to estimate the dose and concentration response of the blood and lung pharmacodynamic endpoints (pAKT/AKT in PRP and BAL cells and 18F-FDG-PET/CT of the whole lung). Estimates and 95% confidence intervals for mean inhibition of each omipalisib dose level relative to placebo at each time point were obtained from the model. PRP pAKT/AKT data were analysed over time on each day of assessment using a repeated measures model, including covariates for treatment, time, baseline (predose) value, and an interaction between time and baseline (predose) value. Estimates and 95% confidence intervals for the mean pAKT/AKT value over time on each day were summarised and plotted. A dose–response model was fitted to the minimum observed pAKT/AKT value for each subject (maximum inhibition) and/or the AUC of the data over time on each day of assessment.
Results
17 participants were enrolled into the study with 13 randomised to omipalisib and four to placebo. Three participants received 0.25 mg omipalisib twice daily, three received 1.0 mg twice daily and seven received 2.0 mg twice daily. One participant who received 2.0 mg twice daily discontinued the study at the request of the sponsor following additional preclinical toxicology data (which was subsequently deemed by the sponsor, ethics committee and MHRA not to impact the benefit/risk assessment of omipalisib in IPF). A CONSORT (Consolidated Standards of Reporting Trials) diagram is provided to illustrate patient flow through the study (figure 1), and subject disposition and demographics are presented in table 1.
Baseline demographics
Population modelling confirmed that the pharmacokinetic profile of omipalisib is best described by a two-compartment, first-order absorption and elimination model. Apparent clearance was 2.2 L·h−1, and apparent volume of distribution in the central and peripheral compartments was 4.9 L and 39.6 L, respectively. The large total volume of distribution indicates potential distribution of omipalisib in body fluid as well as into tissues. A summary of predicted Cmax, Ctrough and AUC0–24 following each dose is shown (table 2).
Summary of population pharmacokinetic model-predicted pharmacokinetic parameters (pharmacokinetic population)
Omipalisib pharmacokinetics in blood on the last day of dosing (figure 2a) show relatively quick absorption with quantifiable concentrations at 30 min post dose. Concentration of omipalisib in blood (immediately before the BAL procedure) and BAL (fluid and cells) were correlated (figure 2c and d).

Omipalisib pharmacokinetics and pharmacodynamics in blood and bronchoalveolar lavage (BAL). a) Left-hand axis shows mean±sem percentage inhibition of pAKT/AKT versus placebo in platelet-rich plasma (PRP) and the right-hand axis shows omipalisib concentration in blood. b) Individual subject pAKT/AKT ratio is presented by dose administered. Ratio to baseline of the pAKT/AKT ratio (weighted mean of 0–6 h time points, 95% CI) in PRP at final dosing day versus omipalisib dose. c) The correlation between the concentration of omipalisib in blood with the concentration of omipalisib in the BAL fluid (trend, 95% CI; individual subject data are illustrated by dose). d) The correlation between the concentration of omipalisib in blood with the concentration of omipalisib in the BAL cells (trend, 95% CI; individual subject data are illustrated by dose). e) Ratio to baseline of pAKT/AKT in BAL cells (log) at mid-study visit versus omipalisib concentration in blood (trend, 95% CI; individual subject data are illustrated by dose). f) Ratio to baseline of BAL PIP3/PIP2 (log) at mid-study visit versus omipalisib concentration in blood (trend, 95% CI; individual subject data are illustrated by dose).
The pharmacokinetic profile of omipalisib on the last day of dosing predicted inhibition of pAKT/AKT in PRP (figure 2a) with a significant dose–response relationship (figure 2b). Increasing omipalisib concentrations in blood showed a trend towards decreased pAKT/AKT (figure 2e) and a significant decrease in PIP3/PIP2 ratios in BAL cells (figure 2f). A maximum effect model (figure 3) provided the best description of the relationship between pAKT/AKT in PRP and blood concentration of omipalisib with a predicted median effective concentration of 74.6 ng·mL−1 (95% CI 20.8–267 ng·mL−1).
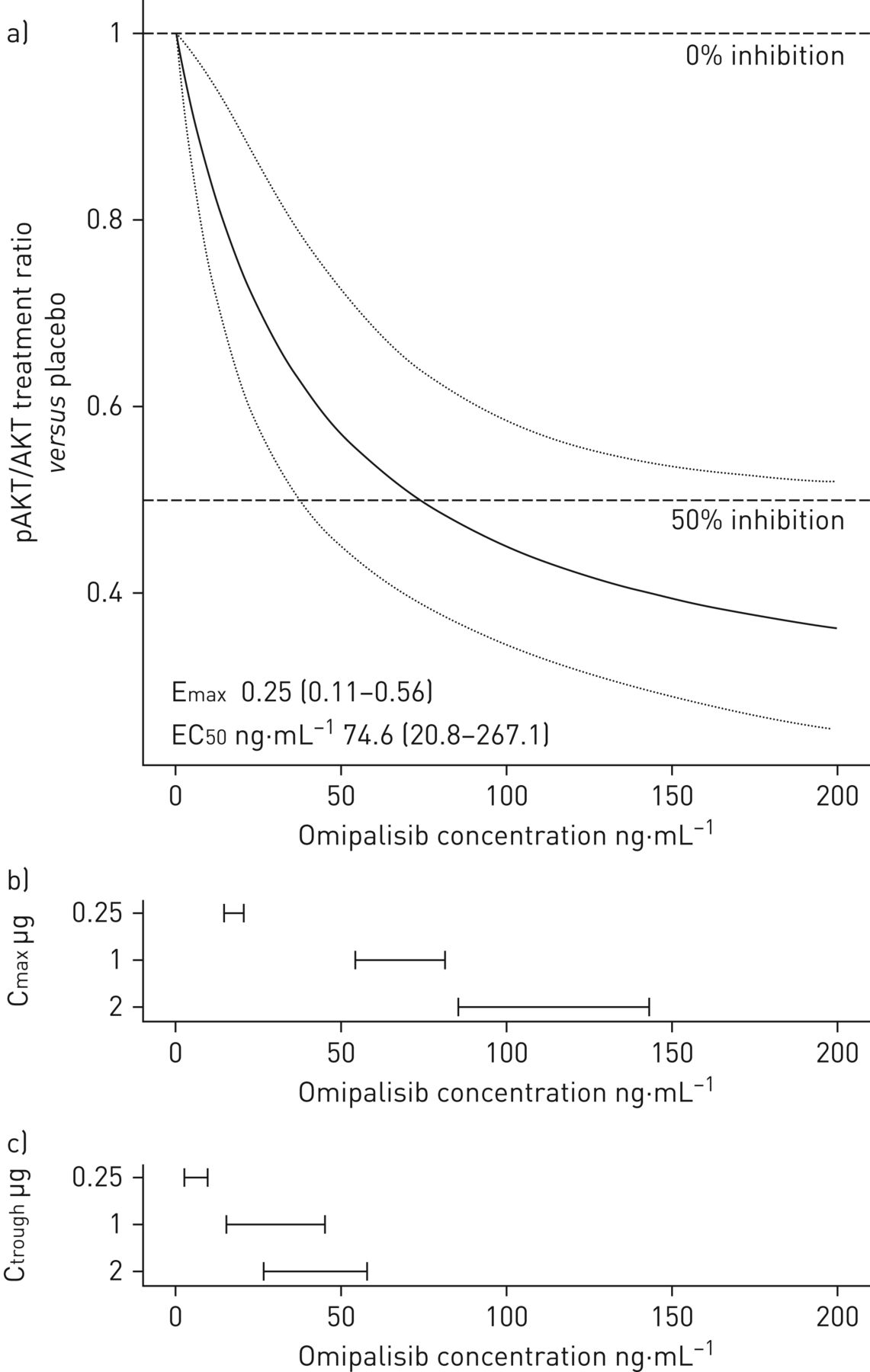
Pharmacokinetic/pharmacodynamic model. a) Fitted maximum effect model of the treatment ratio (versus placebo) in pAKT/AKT on the final day of dosing (adjusted for pAKT/AKT at baseline) versus blood omipalisib concentration (black line and confidence band) (upper panel); the observed range of b) maximum observed concentration (Cmax) and c) predose (trough) concentration at the end of the dosing interval (Ctrough) for the three active treatment doses.
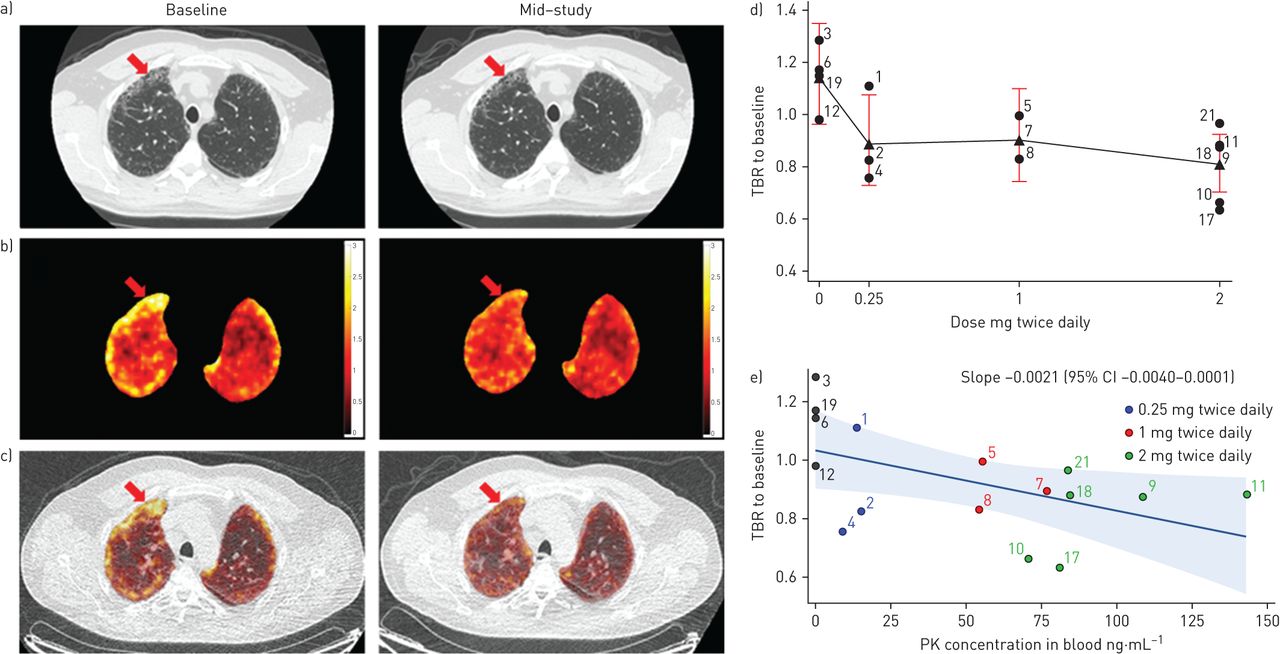
Analysis of whole-lung FDG-PET (SUVmean, SUV99% and Ki did not show an effect of omipalisib with or without air fraction correction. However, a possible regional effect of omipalisib was apparent on visual inspection of images (figure 4a–c) and subsequent re-analysis of the FDG-PET data using TBR revealed a dose- and exposure-dependent reduction TBR (figure 4d and e). Although some of the increased 18F-FDG-PET signal in diseased regions could be explained by reduced air fraction in diseased lung [28, 29], the average lung density assessed from combined CT images did not change between the baseline and mid-study scans, thus confirming that air fraction correction was not necessary for within-subject comparison. Dynamic PET analysis accounts for the component of the signal arising from blood (reducing any effect of changes in blood glucose level due to omipalisib). When combined with air fraction correction dynamic analysis provides the influx rate (Ki) arising only from lung cells. Unfortunately, Ki was found to be too variable to enable reliable interpretation in this small sample size.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of omipalisib on 18F-fluoro-2-deoxy-d-glucose (FDG) positron emission tomography (PET). a) High-resolution computed tomography (HRCT) of the lungs of a representative subject dosed with 2 mg twice daily of omipalisib taken at inspiration with reconstruction diameter of 37.2 cm at baseline and mid-study scan (greyscale window −1350–150 HU); b) PET standardised uptake value (SUV) images of the same subject (before air fraction correction), masked to show lung only; and c) fused PET/computed tomography attenuation correction (CTAC) of the same subject at (left) baseline and (right) mid-study registered to baseline scan. The low-dose CTAC (greyscale window −1350–150 HU) was acquired for attenuation correction with reconstruction diameter of 50 cm. The arrow shows an area of fibrosis that is apparent as honeycombing on the HRCT and that shows high FDG uptake at baseline which is reduced at mid-study. d) The mean (filled triangle), individual subject values (filled circle) and 95% CI of target to background ratio (TBR) of mid-study to baseline versus administered dose and e) the TBR of mid-study to baseline ratio versus omipalisib plasma concentration measured at the mid-study PET visit. Data are presented as individual subject values, regression line and 95% confidence interval. TBR is reported without air fraction correction.
Drug-related adverse events are tabulated (table 3). No treatment-related adverse events were reported in the placebo group. Two (out of three) subjects reported adverse events in the 0.25 mg twice daily group (diarrhoea and nausea), one subject (out of three) reported diarrhoea in the 1 mg twice daily group and four (out of seven) subjects suffered adverse events in the 2 mg twice daily group (hyperglycaemia, lymph node pain, oral pain, rash and rhinitis). No serious adverse events were reported. No adverse events led to early termination of treatment. None of the clinical laboratory, vital signs or ECG abnormalities was clinically significant and none was reported as an adverse event.
Summary of drug-related adverse events (safety population)
A treatment-related decrease in ECG heart rate was apparent, although a consistent dose response was not observed (supplementary figure S1). The decreases in heart rate were not considered to be clinically significant and were not reported as adverse events during the study. There was no significant difference in adjusted mean Leicester Cough Questionnaire or hand-held spirometry (forced expiratory volume in 1 s and FVC) for any treatment group.
Most subjects dosed with 1 mg twice daily or 2 mg twice daily omipalisib had fasting or post-prandial glucose levels above the upper limit of normal and associated with a significant rise in insulin levels (supplementary figure S2). This increase is a class effect that has been observed in other studies of PI3K/mTOR inhibitors [30]. Hyperglycaemia reversed after discontinuation of treatment.
Discussion
This is the first study to adopt an experimental medicine approach to evaluating the pharmacology of a novel therapeutic targeting the PI3K/mTOR pathway in IPF. Using a combination of blood, BAL and imaging biomarkers, this study demonstrates the feasibility of performing intensive experimental studies in IPF. It is hoped that this paradigm of providing robust early evidence of clinical pharmacology may be adopted in the development of novel IPF therapies in the future; thus avoiding unnecessary exposure of large numbers of subjects to ineffective therapies [31, 32]. We have demonstrated a relationship between omipalisib inhibition of PI3K/mTOR in the circulation and in the lungs. Thus, at concentrations that were tolerated, omipalisib inhibits PI3K/mTOR and demonstrates important downstream effects on key intracellular signalling in the fibrotic lung as measured by pAKT, PIP3 and FDG-PET.
The pharmacokinetic of omipalisib had been described in a first-dose-in-humans study in subjects with cancer (www.clinicaltrials.gov identifier number NCT00972686). These data were used to build a pharmacokinetic model that predicted exposure in IPF [11]. The observed pharmacokinetic values from the current study were used to refine the model to more accurately predict pharmacokinetic in this population. This model was supported by extensive in vitro pharmacology exploring the effect of omipalisib on key fibrotic processes, including fibroblast proliferation and TGF-β-induced collagen synthesis [11]. The predicted pharmacologically active dose range for omipalisib, based on in vitro data [11], fell between 0.25 mg twice daily and 2.0 mg twice daily. Pharmacokinetics observed in IPF subjects were consistent with the model, with the chosen doses inhibiting the PI3K/mTOR pathway as predicted.
Class I PI3K catalyses the conversion of PIP2 to PIP3, in turn inducing phosphorylation of the downstream kinase, AKT [12]. Inhibition of AKT phosphorylation by omipalisib has been shown to exert antifibrotic effects in IPF fibroblasts [11]. While it was not feasible to obtain myofibroblasts (which would require repeat open-lung biopsy), we anticipate that inhibition of PI3K in (myo)fibroblasts should have occurred based on effective distribution of omipalisib from the blood into the alveolar compartment together with measurable reduction in PIP3/PIP2 and AKT phosphorylation in BAL cells. A maximum effect model best describes the relationship between systemic pAKT/AKT and the concentration of omipalisib in the blood. Adjusting for blood–protein binding, this model was in agreement with a previously reported in vitro pharmacokinetic/pharmacodynamic model of pAKT in IPF myofibroblasts [11].
FDG-PET was used in this study as a pharmacodynamic endpoint to explore the effect of omipalisib on glucose metabolism in the lungs of IPF participants. Increased FDG uptake in honeycomb areas of IPF lung has previously been described [8] and shown to be reproducible over relatively short periods [9]. The underlying biological processes resulting in increased metabolic activity in IPF lung are unknown. However, numerous cell types in the lung, including structural and immune cells express the GLUT1 receptor necessary for glucose uptake [33, 34] and FDG-PET activity. Furthermore, several studies have demonstrated alterations in glycolysis in both whole IPF lung and in fibroblasts isolated from fibrotic tissue [10, 35]; suggesting that the observed PET signal may reflect a range of abnormal, metabolic processes ongoing within fibrotic lung tissue.
IPF is temporally and spatially heterogeneous, with relatively unaffected areas of lung adjacent to regions of active fibrosis. By measuring uptake of FDG within fibrotic regions in relation to uptake in relatively normal regions (TBR), it was possible to demonstrate that PI3K/mTOR inhibition results in a reduction in metabolic activity. In addition, a significant relationship between reduction in TBR and levels of omipalisib measured in blood (and BAL) of participants was observed. TBR of FDG-PET has recently been shown to predict mortality in IPF [26], with high TBR at baseline correlating with poor prognosis. FDG-PET could therefore potentially represent a valuable, non-invasive, surrogate efficacy measure for use in early clinical development. Further studies are required to understand the long-term relevance of attenuating FDG-PET signal in fibrotic lung.
Analysis of FDG-PET/CT data in the lungs of subjects with IPF is complex, with many confounding factors. These include air- and blood-flow disruption [36], regional differences in tissue density [37] and differences in cell populations compared with healthy lungs [38]. Metabolic changes due to omipalisib were observed using TBR; a technique that expresses FDG-PET signal as a ratio between uptake in selected fibrotic and relatively non-fibrotic regions. Thus, TBR uses an internal reference which compensates for some of the potential confounders when interpreting metabolic activity in IPF lung. This study contributes not only to understanding of PI3K/mTOR inhibition in IPF, but also to technical aspects of using FDG-PET as an endpoint in fibrotic lung disease.
There are some potential limitations of this study that should be considered. It was designed as an experimental medicine study to understand the pharmacological and biological relevance of attenuating PI3K signalling with a potent small-molecule inhibitor in IPF lung. As such, only small numbers of subjects were enrolled and dosed with omipalisib for a short period of time. This permitted a clear understanding of the pharmacological effect of different concentrations of omipalisib, but did not permit any assessment of efficacy. Furthermore, although the drug was continued by all subjects for the duration of the study, longer-term tolerability of omipalisib in IPF remains to be determined. Another potential limitation is that participants underwent PET/CT followed by BAL twice during a short time frame. Theoretically, residual effects of the baseline BAL may have impacted the mid-study PET/CT; however, the study was placebo controlled and so any effect of BAL should have been consistent across groups. Most of the endpoints were analysed prior to unblinding of the data, with the exception of TBR analysis of FDG-PET data which was conducted after unblinding of the study. To mitigate any potential bias in interpretation of scans, all reference to study treatment was removed from FDG-PET/CT scans prior to post hoc analysis. A strength of this study is the clear demonstration of PI3K target engagement in fibrotic lung with demonstrable effects on lung metabolism. This suggests that it is feasible to inhibit PI3K in IPF subjects, but later-phase trials will be required to determine any clinically relevant effect on fibrosis.
Omipalisib was reasonably well tolerated in this short-term dose escalation study. It was anticipated based on oncology experience that diarrhoea could be a dose-limiting adverse event [39, 40]. Although there were a few cases of diarrhoea in the active group, these events were mild or moderate in intensity, were never of concern and did not lead to stopping of treatment. It is worth noting that adverse events related to the gastrointestinal tract are observed in IPF subjects treated with pirfenidone [3] and nintedanib [2]. The observed increases in insulin and glucose suggest a degree of insulin resistance in some subjects; this was expected, based on the known pharmacology of PI3K/mTOR inhibition [30]. In addition, PI3K inhibition appeared to be associated with a mild, but clinically nonsignificant, negative chronotropic effect.
In conclusion, we have shown that orally dosed omipalisib exerts measurable dose- and exposure-dependent inhibition of PI3K/mTOR pathway in the systemic circulation and lungs of individuals with IPF. Omipalisib also reduces aberrant glucose signalling in fibrotic regions of IPF lungs as measured by FDG-PET. These data support further assessment of the PI3K/mTOR pathway as a target for new treatments for IPF. Inhibitors with improved selectivity may minimise or avoid potential side-effects of broader PI3K/mTOR inhibition while providing similar potential therapeutic benefit.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figures S1 and S2 ERJ-01992-2018_Supplement
Acknowledgements
The authors would like to thank the wider team members who contributed to the successful conduct of this study. The GSK team consisting of Maria Costa, Jane Bentley, Nadia Garman, Tom Lumbers, Stuart Kendrick, Val Morrison, Tina Sawhney, Vikki Barratt, Simon Parry, Arthur Kang'ombe and Jane Gilbert. The Brompton team consisting of Shaun Kingston and Una Fraser. The UCL team consisting of Vesna Cuplov, Elise Emond, Raymond Endozo, Francesco Fraioli, Brian Hutton, Lynn Millner, Benjamin Thomas, Robert Shortman Manuela Plate and Linda Franklin. In addition, we would like to thank Jonathan Clark (Babraham Institute) for his contribution to the PIP3/PIP2 assays.
Footnotes
This article has supplementary material available from erj.ersjournals.com
This study is registered at clinicaltrials.gov with identifier number NCT01725139. The results summary for this study (NCT01725139 or IPF116711) is available on the GSK Clinical Study Register at www.gsk-clinicalstudyregister.com. Anonymised patient-level data will be made available to independent researchers, subject to review by an independent panel, at www.clinicalstudydatarequest.com within 6 months of publication. To protect the privacy of patients and individuals involved in our studies, GSK does not publicly disclose patient-level data.
Author contributions: P.T. Lukey, S.A. Harrison, S. Yang, J.K. Simpson, P. Bareille, A.M. Groves, R.P. Marshall and T.M. Maher made substantial contributions to the conception and design of the study. Y. Man, B.F. Holman, A. Rashidnasab, G. Azzopardi, M. Grayer, LP, H.V. Woodcock, R. Toshner, P. Saunders, P.L. Molyneaux, K. Thielemans, F.J. Wilson, P.F. Mercer, R.C. Chambers and W.A. Fahy made substantial contributions to the acquisition, analysis, or interpretation of data for the study. All the authors contributed to drafting the manuscript or revising it critically for important intellectual content; all authors gave final approval of the version to be published; and all authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest: P.T. Lukey was a GSK employee at the time of the study and is still a shareholder. P.T. Lukey now works or has worked as an independent consultant to GSK R&D, the Francis Crick Institute, Syncona, Mereo BioPharma, Peptinnovate, BerGenBio, Morphic Therapeutics and LifT BioSciences.
Conflict of interest: S.A. Harrison reports that the study was funded by GlaxoSmithKline; and is an employee of and holds shares in GlaxoSmithKline, outside the submitted work.
Conflict of interest: S. Yang reports that the study was funded by GlaxoSmithKline; and is an employee of and holds shares in GlaxoSmithKline, outside the submitted work.
Conflict of interest: Y. Man has nothing to disclose.
Conflict of interest: B.F. Holman reports grants from EPSRC, during the conduct of the study.
Conflict of interest: A. Rashidnasab reports grants from GSK, during the conduct of the study.
Conflict of interest: G. Azzopardi has nothing to disclose.
Conflict of interest: M. Grayer reports that the study was funded by GlaxoSmithKline; and received personal fees as a contractor on assignment at GSK to complete statistical analyses, from GlaxoSmithKline, outside the submitted work.
Conflict of interest: J.K. Simpson is an employee of and holds shares in GlaxoSmithKline.
Conflict of interest: P. Bareille is an employee of and holds shares in GlaxoSmithKline.
Conflict of interest: L. Paul has nothing to disclose.
Conflict of interest: H.V. Woodcock reports grants from GlaxoSmithKline, during the conduct of the study.
Conflict of interest: R. Toshner has nothing to disclose.
Conflict of interest: P. Saunders has nothing to disclose.
Conflict of interest: P.L. Molyneaux has nothing to disclose.
Conflict of interest: K. Thielemans reports grants from GSK, during the conduct of the study; grants from GE Healthcare, outside the submitted work.
Conflict of interest: F.J. Wilson is an employee of and holds shares in GlaxoSmithKline.
Conflict of interest: P.F. Mercer was funded through a collaborative framework agreement with GlaxoSmithKline.
Conflict of interest: R.C. Chambers reports grants from GlaxoSmithKline (GSK), during the conduct of the study; and R.C. Chambers’ spouse is an employee of GSK.
Conflict of interest: A.M. Groves has nothing to disclose.
Conflict of interest: W.A. Fahy is an employee of and holds shares in GlaxoSmithKline.
Conflict of interest: R.P. Marshall is an employee of and holds shares in GlaxoSmithKline, outside the submitted work.
Conflict of interest: T.M. Maher has, via his institution, received industry-academic funding from GlaxoSmithKline R&D and UCB and has received consultancy or speaker's fees from Apellis, Astra Zeneca, aTyr Pharma, Bayer, Biogen Idec, Boehringer Ingelheim, Galapagos, GlaxoSmithKline R&D, ProMetic, Roche, Sanumed and UCB.
Support statement: GSK R&D sponsored and funded the study. T.M. Maher is supported by an NIHR Clinician Scientist Fellowship (NIHR Ref: CS-2013-13-017) and a British Lung Foundation Chair in Respiratory Research (C17-3). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received August 2, 2018.
- Accepted December 11, 2018.
- Copyright ©ERS 2019
References










