





Abstract
TELOS compared budesonide (BD)/formoterol fumarate dihydrate (FF) metered dose inhaler (BFF MDI), formulated using innovative co-suspension delivery technology that enables consistent aerosol performance, with its monocomponents and budesonide/formoterol fumarate dihydrate dry powder inhaler (DPI) in patients with moderate to very severe chronic obstructive pulmonary disease (COPD), without a requirement for an exacerbation history.
In this phase III, double-blind, parallel-group, 24-week study (NCT02766608), patients were randomised to BFF MDI 320/10 µg (n=664), BFF MDI 160/10 µg (n=649), FF MDI 10 µg (n=648), BD MDI 320 µg (n=209) or open-label budesonide/formoterol DPI 400/12 µg (n=219). Primary end-points were change from baseline in morning pre-dose trough forced expiratory volume in 1 s (FEV1) and FEV1 area under the curve from 0–4 h (AUC0–4). Time to first and rate of moderate/severe exacerbations were assessed.
BFF MDI 320/10 µg improved pre-dose trough FEV1 versus FF MDI (least squares mean (LSM) 39 mL; p=0.0018), and BFF MDI 320/10 µg and 160/10 µg improved FEV1 AUC0–4 versus BD MDI (LSM 173 mL and 157 mL, respectively; both p<0.0001) at week 24. BFF MDI 320/10 µg and 160/10 µg improved time to first and rate of moderate/severe exacerbations versus FF MDI. Treatments were well tolerated, with pneumonia incidence ranging from 0.5–1.4%.
BFF MDI improved lung function versus monocomponents and exacerbations versus FF MDI in patients with moderate to very severe COPD.
Abstract
TELOS: co-suspension delivery technology budesonide/formoterol fumarate dihydrate in a metered dose inhaler improved lung function and time to first and rate of exacerbations versus monocomponents in patients with moderate to very severe COPD http://ow.ly/ffWo30lrJL6
Introduction
Recommendations for the use of inhaled corticosteroids (ICS) in the treatment of chronic obstructive pulmonary disease (COPD) have changed over recent years. Dual ICS/long-acting β2-agonist (LABA) therapy is currently recommended as an optional treatment for patients with a high risk of COPD exacerbations [1]. Previous studies have shown that combined ICS/LABA treatment was more effective than ICS or LABA monotherapy or placebo in improving lung function, dyspnoea and health status, and in reducing the rate of exacerbations [2–11]. Previous findings also suggest that patients with higher blood eosinophil levels may benefit more from therapies that include an ICS with regard to reducing the risk of exacerbations than those with lower eosinophil levels [12–14].
The ICS/LABA budesonide (BD)/formoterol fumarate dihydrate (FF) metered dose inhaler (BFF MDI), formulated using innovative co-suspension delivery technology, is currently being developed as a maintenance treatment for patients with COPD. Co-suspension delivery technology enables reliable aerosol performance and consistent drug delivery, even in the presence of simulated patient-handling errors, unlike drug crystal-only suspensions [15, 16]. In a scintigraphy study, co-suspension delivery technology MDIs have also been shown to provide effective delivery throughout the whole lung [17].
The TELOS study examined the efficacy and safety of 24 weeks of treatment with two doses of BFF MDI (320/10 µg and 160/10 µg) compared with BD MDI 320 µg, FF MDI 10 µg and budesonide/formoterol fumarate dihydrate dry powder inhaler (DPI) 400/12 µg in symptomatic patients (COPD Assessment Test (CAT) score ≥10) with moderate to very severe COPD, without a requirement for a history of exacerbations. The relationship between baseline blood eosinophil count and improvements in lung function and COPD exacerbations was also investigated. Following a phase IIb BFF MDI dose-ranging study [18], two doses of BFF MDI were included to investigate whether there was a dose ordering effect of budesonide on therapeutic response.
Methods
Study population
Eligible patients were current or former smokers (≥10 pack-years), 40–80 years of age, with an established clinical history of COPD, as defined by American Thoracic Society/European Respiratory Society [19] or locally applicable guidelines. Patients had to be symptomatic (CAT score ≥10) despite treatment with one or more inhaled bronchodilator as COPD maintenance therapy for ≥6 weeks prior to screening. There was no requirement for prior COPD exacerbations.
Patients were required to have a forced expiratory volume in 1 s (FEV1) to forced vital capacity (FVC) ratio <0.70 and FEV1 <80% of predicted normal value at the first screening visit, and a post-bronchodilator FEV1/FVC ratio <0.70 and post bronchodilator FEV1 ≥30% but <80% of predicted normal value at the second screening visit. If the post-bronchodilator FEV1 was <30% of predicted normal at the second screening visit, then FEV1 must have been ≥750 mL. At randomisation, the average of the 30-min and 60-min pre-dose FEV1 assessments must have been <80% of predicted normal value.
Exclusion criteria included treatment with ICS, long-acting muscarinic antagonist and LABA as triple maintenance therapy in the past 30 days, a diagnosis of a respiratory disorder other than COPD (including a current diagnosis of asthma) or COPD due to α1-antitrypsin deficiency. Patients who were hospitalised within 6 weeks, or who had been treated with systemic corticosteroids and/or antibiotics for COPD within 4 weeks prior to screening were excluded, as were patients with other clinically relevant diseases that could impact on study findings. Additional exclusion criteria included a change in smoking status within 6 weeks prior to or during screening, requirement for long-term oxygen therapy (≥15 h per day) or the need to use a spacer device with an MDI. Patients were required to demonstrate an acceptable MDI administration technique, as assessed by study site personnel.
Study design and treatment
TELOS, a randomised, double-blind, parallel-group, 24-week, phase III study, was conducted at 253 sites across seven countries (Canada, Czech Republic, Germany, Hungary, Poland, Russia and the USA) between June 2016 and November 2017. Study site personnel randomised patients 3:3:3:1:1 using an Interactive Web Response System to treatment with BFF MDI 320/10 µg, BFF MDI 160/10 µg, FF MDI 10 µg, BD MDI 320 µg or open-label budesonide/formoterol DPI 400/12 µg (Symbicort® Turbohaler®; AstraZeneca UK Limited, Luton, UK). All treatments were administered as two inhalations twice daily for 24 weeks. Formoterol fumarate dihydrate 10 µg is equivalent to formoterol fumarate 9.6 µg. Budesonide/formoterol DPI was given as two inhalations of 200/6 µg, equivalent to a total delivered dose of 320/9 µg. BFF MDI, FF MDI and BD MDI were given in identical MDI devices, and all participants, investigators and study personnel were blinded to the MDI treatment assigned.
Eligible patients were required to discontinue COPD treatment with prohibited medications during screening and for the duration of the study. Patients who received an ICS as part of their inhaled maintenance therapy continued the ICS component during the screening period. ICS were discontinued at randomisation. Salbutamol sulfate (albuterol sulfate; Ventolin® HFA; GlaxoSmithKline, Research Triangle Park, NC, USA) was used as rescue medication throughout the study as needed for symptoms. Xanthine- or caffeine-containing foods or beverages and caffeine-containing medications were prohibited for ≥6 h prior to, and for the duration of, each in-clinic study visit.
Randomisation was stratified by reversibility to salbutamol (yes/no), post-bronchodilator FEV1 at the second screening visit (<50%, or 50% to <80% predicted), baseline blood eosinophil count (<150 or ≥150 cells per mm3; enrolment was targeted to achieve a 1:2 ratio of the number of patients with blood eosinophil levels <150 cells per mm3 relative to ≥150 cells per mm3) and country.
The study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation/Good Clinical Practice and applicable regulatory requirements. The study protocol and informed consent form were reviewed and approved by the appropriate independent ethics committee or institutional review board, and patients provided written informed consent prior to screening. Protocol amendments pertaining to study end-points or data analyses are shown in supplementary table S1. This study is registered with ClinicalTrials.gov, number NCT02766608.
Efficacy end-points and assessments
Two registration approaches, US and EU, were used in the study. Primary and secondary end-points, and the time frames of each, differed according to regional regulatory requirements.
For the US approach, change from baseline in morning pre-dose trough FEV1 (BFF MDI versus FF MDI) and change from baseline in FEV1 area under the curve from 0–4 h (AUC0–4) (BFF MDI versus BD MDI), both at week 24, analysed using the efficacy estimand were primary end-points. The same end-points analysed using the attributable estimand were secondary end-points. Change from baseline in morning pre-dose trough FEV1 at week 24 (BFF MDI versus BD MDI; efficacy estimand) was a secondary end-point. Other secondary end-points in the US approach were peak change from baseline in FEV1 at week 24 (BFF MDI versus BD MDI), time to first moderate/severe COPD exacerbation (BFF MDI versus FF MDI), change from baseline in average daily rescue salbutamol use over 24 weeks (BFF MDI versus BD MDI), percentage of patients with an improvement in St George's Respiratory Questionnaire (SGRQ) total score ≥4 at week 24 (BFF MDI versus BD MDI and FF MDI) and time to onset of action as assessed by FEV1 on day 1 (BFF MDI versus BD MDI). Additional end-points included the rate of moderate/severe COPD exacerbations. End-points for the EU approach, which generally analysed end-points over 24 weeks (rather than at week 24), are described in the supplementary materials.
COPD exacerbations are defined in the supplementary materials and were considered moderate if they resulted in use of systemic corticosteroids and/or antibiotics for ≥3 days, and severe if they resulted in COPD-related hospitalisation or death. Pre-specified subgroup analysis of the two primary end-points and the rate of moderate/severe COPD exacerbations was performed based on a baseline blood eosinophil count of <150 cells per mm3 and ≥150 cells per mm3. Blood eosinophil counts were measured at screening visit 1 and at randomisation, and the mean of the non-missing values was recorded as the baseline blood eosinophil count. Further details on efficacy assessments are included in the supplementary materials.
Safety evaluation
Safety assessments included physical examinations, vital signs assessments, 12-lead ECG and clinical laboratory testing. Adverse events were monitored throughout the study. Predefined criteria for identifying and reporting pneumonia and major adverse cardiovascular events (MACE) were implemented, which are described in the supplementary materials. An external, independent clinical end-point committee reviewed all adverse events reported as pneumonia and adverse events that potentially met MACE criteria.
Statistical analysis
The intent-to-treat (ITT) and safety populations were defined as all patients who were randomised and received study treatment. The ITT population was analysed according to the treatment patients were randomised to and the safety population was analysed by the treatment received. The modified intent-to-treat (mITT) population included ITT population data that were obtained post-randomisation but prior to discontinuation from treatment.
The primary estimand was the efficacy estimand, which was defined as the effect of the randomised treatment in all patients assuming continuation of randomised treatments for the duration of the study regardless of actual compliance. The primary analysis for the efficacy estimand was conducted using the mITT population. An attributable estimand was defined as the effect of treatment in patients attributable to the randomised treatment, and considered treatment discontinuation due to lack of efficacy or tolerability as a bad outcome. Analyses of the attributable estimand were conducted using the mITT population, but with imputation of data following treatment discontinuation.
The two primary end-points were analysed using a repeated measures linear mixed model. The model included treatment, visit (and treatment by visit interaction) and ICS use at screening as categorical covariates, and baseline FEV1, baseline blood eosinophil count and percent reversibility to salbutamol as continuous covariates. An unstructured covariance matrix was used to model correlation within a patient. Two-sided p-values and point estimates with two-sided 95% confidence intervals were produced for each treatment difference of interest.
The US and EU approaches controlled the type I error for the primary and secondary analyses with a combination of sequential and Hochberg methods (US approach; supplementary figure S1). Comparisons were reported as nominally significant if they were p<0.05 but failed statistical significance due to the type I error control strategy. The statistical analysis methods for secondary end-points, subgroup analyses, type I error control for the EU approach, non-inferiority comparisons and power calculations are detailed in the supplementary materials.
Results
Study population
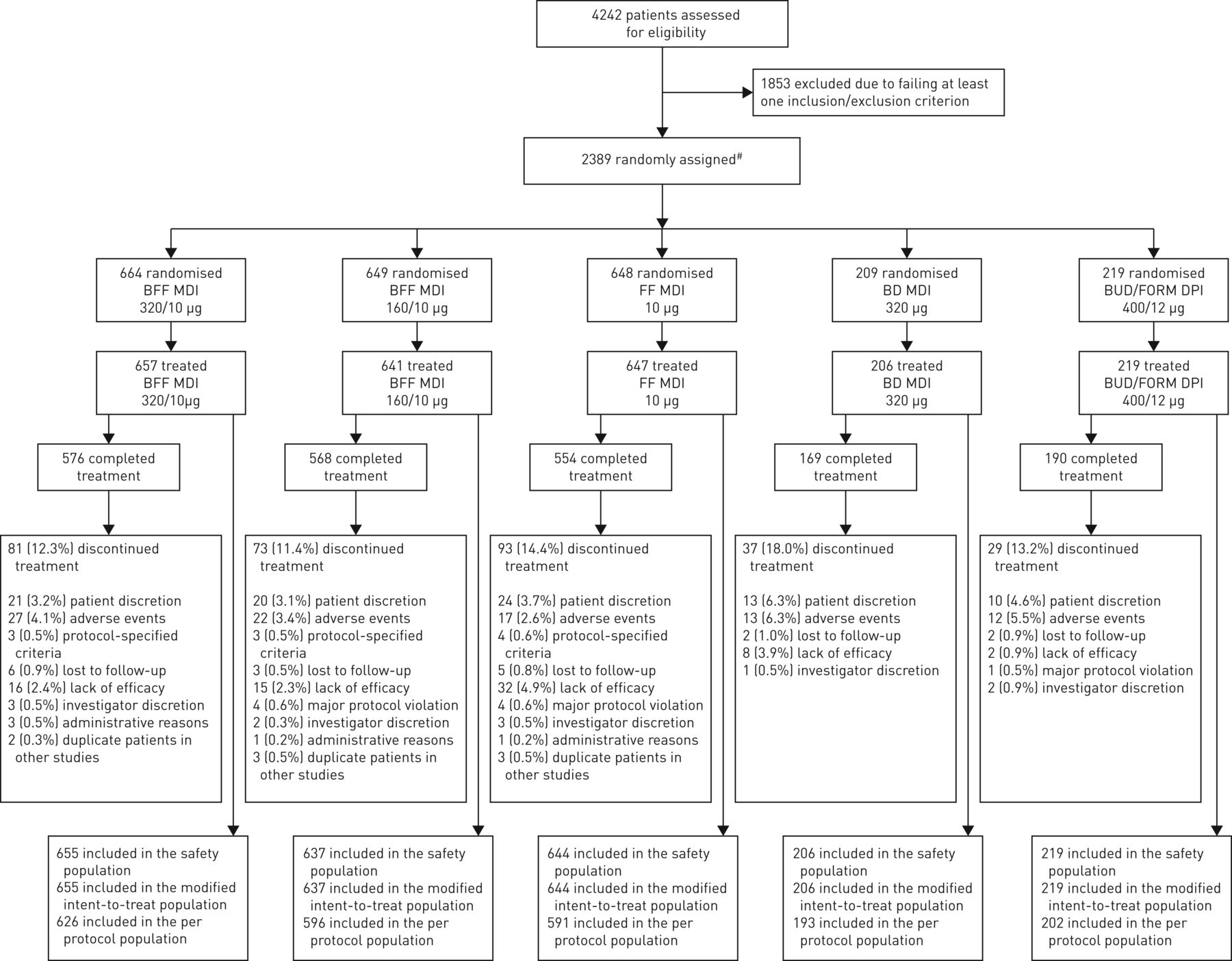
Overall, 4242 patients were screened, and 2389 patients were randomised. Of these, 2370 patients (99.2%) received treatment and 2057 (86.1%) completed 24 weeks of treatment. The mITT and safety populations included 2361 patients (98.8%) (figure 1).

Patient disposition. BD: budesonide; BFF: budesonide/formoterol fumarate dihydrate; BUD/FORM DPI: budesonide/formoterol dry powder inhaler; FF: formoterol fumarate dihydrate; MDI: metered dose inhaler. #: 19 patients who were randomised were not treated: 14 did not meet baseline stability criteria; one due to chronic obstructive pulmonary disease diagnosis; one due to eDiary compliance; one due to prohibited medication; one was a screen failure (non-specific); and one was a “mistake”. One patient in the BFF MDI 160/10 µg group was identified as a duplicate patient and excluded from the analysis populations, but completed treatment.
Most patients in the mITT population were white (96.6%) and male (60.5%), with a mean±sd age of 64.3±7.7 years, smoking history of 44.9±23.0 pack-years and post-bronchodilator FEV1 of 52.93±13.01% of predicted. Overall, 49.0% of patients were using ICS at screening; 72.9% of patients had no history of moderate/severe COPD exacerbations in the year prior to screening (table 1), and 90.0% of patients had ≤1 moderate/severe COPD exacerbation, not requiring hospitalisation. Per study design, 34.3% of patients had a baseline blood eosinophil count <150 cells per mm3 and 65.7% of patients had a baseline eosinophil count ≥150 cells per mm3 (table 1; supplementary figure S2). Demographic characteristics, including baseline eosinophil count and exacerbation history, were balanced across the treatment groups (table 1).
Baseline demographics and clinical characteristics (safety/modified intent-to-treat population)
Primary end-points
BFF MDI 320/10 µg statistically significantly improved change from baseline in morning pre-dose trough FEV1 at week 24 compared with FF MDI (demonstrating the ICS effect; least squares mean (LSM) 39 mL; p=0.0018), whereas numerical improvements were observed for the comparison of BFF MDI 160/10 µg with FF MDI (LSM 20 mL; p=0.1132) (figure 2a; table 2). BFF MDI 320/10 µg and 160/10 µg statistically significantly improved FEV1 AUC0–4 at week 24 compared with BD MDI (demonstrating the LABA effect; LSM 173 mL and 157 mL, respectively; both p<0.0001) (figure 2b; table 2). Analyses of the two primary end-points at week 24 using the attributable estimand (an analysis that adjusts for the potential bias introduced by data missing due to lack of efficacy or tolerability) were comparable with those using the efficacy estimand (table 2).

Primary efficacy end-points (efficacy estimand; modified intent-to-treat population). a) Adjusted mean change from baseline in morning pre-dose trough forced expiratory volume in 1 s (FEV1) over time. b) Adjusted mean change from baseline in FEV1 area under the curve from 0–4 h (AUC0–4) over time. Data are presented as least squares means with standard errors. BD: budesonide; BFF: budesonide/formoterol fumarate dihydrate; BUD/FORM DPI: budesonide/formoterol dry powder inhaler; FF: formoterol fumarate dihydrate; MDI: metered dose inhaler.
Primary, secondary and other efficacy end-points (efficacy estimand, unless stated otherwise; modified intent-to-treat population)
Results over 24 weeks were similar to those at week 24, and also showed that BFF MDI 320/10 µg was non-inferior to budesonide/formoterol DPI for the two EU primary end-points (supplementary table S2).
Secondary lung function end-points
Both doses of BFF MDI nominally significantly improved morning pre-dose trough FEV1 at week 24 versus BD MDI (figure 2a; table 2). BFF MDI 320/10 µg statistically significantly improved peak change from baseline in FEV1 at week 24 versus BD MDI, with findings for BFF MDI 160/10 µg versus BD MDI reaching nominal significance (table 2). Onset of action for both doses of BFF MDI occurred within 5 min (table 2).
Exacerbations
Overall, 16.9% of patients with BFF MDI 320/10 µg, 19.9% of patients with BFF MDI 160/10 µg, 23.3% of patients with FF MDI, 18.9% of patients with BD MDI, and 14.6% of patients with open-label budesonide/formoterol DPI experienced a moderate/severe COPD exacerbation. BFF MDI 320/10 µg statistically significantly improved time to first moderate/severe COPD exacerbations versus FF MDI (hazard ratio (HR) 0.675, 95% CI 0.528, 0.863; p=0.0017), with the comparison of BFF MDI 160/10 µg versus FF MDI showing nominal significance (HR 0.771, 95% CI 0.608, 0.977; p=0.0310) (table 2; supplementary figure S3). The adjusted rate of moderate/severe COPD exacerbations per year was 0.44 for BFF MDI 320/10 µg, 0.50 for BFF MDI 160/10 µg, 0.69 for FF MDI, 0.65 for BD MDI, and 0.32 for open-label budesonide/formoterol DPI. BFF MDI 320/10 µg and 160/10 µg nominally significantly reduced the rate of moderate/severe COPD exacerbations by 37% and 28% versus FF MDI, respectively (table 2).
Symptom end-points
BFF MDI 320/10 µg statistically significantly improved average daily rescue salbutamol use over 24 weeks versus BD MDI, and the SGRQ responder rates (improvement in SGRQ total score ≥4 units) at week 24 versus FF MDI (table 2).
Eosinophil subgroups
Treatment differences in change from baseline in morning pre-dose trough FEV1 at week 24 between BFF MDI 320/10 µg and 160/10 µg and FF MDI were 48 mL (95% CI 17, 79 mL) and 27 mL (95% CI −5, 59 mL), respectively, in the ≥150 cells per mm3 subgroup, and 21 mL (95% CI −18, 60 mL) and 7 mL (95% CI −31, 45 mL), respectively, in the <150 cells per mm3 subgroup. Comparisons of BFF MDI versus BD MDI did not show a greater treatment difference in the ≥150 cells per mm3 subgroup (figure 3a). Treatment differences in change from baseline in FEV1 AUC0–4 at week 24 between BFF MDI (both doses) and both monocomponents were comparable in both eosinophil subgroups (figure 3b).

Forest plots of primary lung function end-points by baseline eosinophil count (efficacy estimand; modified intent-to-treat (mITT) population; US approach). a) Change from baseline in morning pre-dose trough forced expiratory volume in 1 s (FEV1) at week 24. b) Change from baseline in FEV1 area under the curve from 0–4 h at week 24. Data are presented as least squares mean treatment differences with 95% confidence intervals. mITT population: <150 cells per mm3, n=810; ≥150 cells per mm3, n=1551; all patients, n=2361. BD: budesonide; BFF: budesonide/formoterol fumarate dihydrate; FF: formoterol fumarate dihydrate; MDI: metered dose inhaler.
Improvements in the rate of moderate/severe COPD exacerbations for BFF MDI 320/10 µg and 160/10 µg versus both monocomponents were observed in both eosinophil subgroups. The rate ratio for BFF MDI 320/10 µg versus FF MDI and BD MDI was 0.57 (95% CI 0.41, 0.78) and 0.63 (95% CI 0.40, 0.98), respectively, for the ≥150 cells per mm3 subgroup, and 0.78 (95% CI 0.50, 1.20) and 0.82 (95% CI 0.43, 1.59), respectively, for the <150 cells per mm3 subgroup, with little difference between the eosinophil subgroups with BFF MDI 160/10 µg (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forest plots of rate of moderate/severe chronic obstructive pulmonary disease exacerbations by baseline eosinophil count (efficacy estimand; modified intent-to-treat (mITT) population; US and EU approach). Data are presented as rate ratios with 95% confidence intervals. mITT population: <150 cells per mm3, n=810; ≥150 cells per mm3, n=1551; all patients, n=2361. BD: budesonide; BFF: budesonide/formoterol fumarate dihydrate; FF: formoterol fumarate dihydrate; MDI: metered dose inhaler.
Safety
The incidence of treatment-emergent adverse events (TEAEs) or TEAEs leading to study drug discontinuation was generally similar across treatment groups, with the incidence of serious TEAEs somewhat higher in the FF MDI group than in the BD MDI and BFF MDI groups (tables 3 and 4). The incidence of adjudicated pneumonia was low in all treatment groups, with an incidence of 0.5% with BD MDI, 0.8% with BFF MDI 320/10 µg, 1.1% with BFF MDI 160/10 µg and 1.4% with FF MDI and budesonide/formoterol DPI, with no dose response observed for BFF MDI (table 3). There were a total of 12 deaths during the study (on-treatment and post-treatment), with an incidence ranging from 0–1.4% across treatment groups, none of which were considered treatment-related (table 3).
Summary of treatment-emergent adverse events (TEAEs) (safety population)
Treatment-emergent adverse events (TEAEs) occurring in ≥2% of patients in any treatment group (safety population)
Discussion
This study in symptomatic patients (CAT score ≥10) with moderate to very severe COPD showed that treatment with BFF MDI 320/10 µg resulted in significant improvements for all pre-specified primary and secondary efficacy comparisons for the US statistical approach, with the exception of SGRQ responder rates at week 24 versus BD MDI, which showed numerical improvements. While improvements were also observed with BFF MDI 160/10 µg, the primary comparison for morning trough FEV1 versus FF MDI did not reach statistical significance. A dose response favouring BFF MDI 320/10 µg was observed for all lung function and exacerbation end-points.
Our study also demonstrated non-inferiority of BFF MDI 320/10 µg, formulated using co-suspension delivery technology, compared to the approved budesonide/formoterol DPI formulation [20]. Choosing an appropriate inhaled drug delivery device for a patient depends on several factors, with each device having its own advantages and challenges. To effectively use a DPI, patients with COPD must be able to generate a sufficient peak inspiratory flow rate to allow the therapy to be inhaled and deposited in the lower respiratory tract [21]. Therefore, an MDI device may be preferable in patients with an inspiratory flow rate that is suboptimal for a DPI. Furthermore, rescue medications are commonly available in MDI devices, and patients with COPD who used multiple devices had more favourable clinical outcomes if these required similar inhalation techniques rather than different techniques [22].
The improvement in change from baseline in morning pre-dose trough FEV1 at week 24 for BFF MDI 320/10 µg versus FF MDI (39 mL) was consistent with the magnitude of improvements observed for approved budesonide/formoterol formulations compared with formoterol alone in 6-month and 12-month studies [4, 8, 23], and also with 6-month studies comparing the ICS/LABA combination fluticasone furoate/vilanterol with vilanterol alone [24, 25].
Overall, adding an ICS to a LABA would be expected to have a greater impact on exacerbations than on lung function outcomes. A recent systematic review reported that patients rate a reduction in exacerbations or hospitalisation due to exacerbations as the most important clinical outcome from COPD therapy [26]. The potential influence of the study design on exacerbation rates is important to consider when interpreting data from trials such as this, and also IMPACT, for which it has been suggested that ICS withdrawal at the end of the screening period could influence the exacerbation rates reported [27]. However, in our study, the time to first moderate/severe COPD exacerbation data showed a cumulative incidence of exacerbations for all therapies that diverged over time, not only in the first 4 weeks (supplementary figure S3), indicating that findings were not only driven by ICS withdrawal after randomisation. Furthermore, it is interesting to note that both doses of BFF MDI improved exacerbation outcomes versus FF MDI despite the study population not being enriched for patients with a history of exacerbations; the majority (73%) had zero exacerbations and 22% had one exacerbation in the prior year. This suggests that ICS/LABA therapy may provide additional benefits compared to LABA monotherapy for some patients with COPD, independent of their recent exacerbation history.
It has been proposed that individualised treatment based on a patient's blood eosinophil levels, along with other respiratory criteria, may be possible [28]. We used baseline blood eosinophil levels of <150 cells per mm3 and ≥150 cells per mm3 to prospectively explore the relationship between eosinophils and improvements in lung function and reductions in COPD exacerbations. Whether a simple threshold may ultimately be used clinically for stratifying patient treatment remains uncertain. However, our study showed trends towards greater lung function and exacerbation benefits in patients with higher blood eosinophil levels, which is consistent with previous studies [12–14, 29]. Further studies are required to determine if blood eosinophil levels can predict treatment responses, and at which thresholds.
While there was a dose response for efficacy with BFF MDI, there was no evidence for a dose response for safety. Importantly, the incidence of adjudicated pneumonia was low among all treatment groups (0.5–1.4%), with the lowest incidences observed with BFF MDI 320/10 µg and BD MDI. The safety profile of BFF MDI was generally similar to that of budesonide/formoterol DPI 400/12 μg, a product with a well-established safety profile [20].
A limitation of this study was the 6-month treatment period, which may not be considered an optimal duration for a study investigating exacerbation rates. However, all four seasons were well represented during the treatment period since the study enrolled patients over 1 year, and the secondary end-point of time to first moderate/severe COPD exacerbation does not require a study to be of a particular duration, since only the first exacerbation is used in the analysis. This end-point showed significant improvements for the comparison of BFF MDI versus FF MDI that were also dose-dependent. In addition, a 6-month treatment period can reduce patient dropout compared to a longer study duration, which may result in the underestimation of exacerbation rates. Notably, the 6-month RISE study, which evaluated a different formulation of budesonide/formoterol MDI, had a primary end-point of the annual rate of COPD exacerbations, and showed comparable results to our study [8]. The lack of patients with asthma−COPD overlap syndrome could be considered a potential limitation of this study, as patients with a current diagnosis of asthma were excluded. However, focusing on patients with COPD ensured that the impact of ICS shown in this study was not due to the effects of ICS in patients with asthma.
Conclusions
Overall, the study demonstrated that BFF MDI 320/10 µg and 160/10 µg effectively improved lung function relative to ICS monotherapy and reduced COPD exacerbations relative to LABA monotherapy in patients with moderate to very severe COPD. A dose response favouring BFF MDI 320/10 µg was observed for all lung function and exacerbation end-points versus both monocomponents. However, there was no dose response observed for safety outcomes, including for pneumonia which had a lower incidence for BFF MDI 320/10 µg relative to BFF MDI 160/10 µg. Finally, consistent with previous studies, there was a relationship between eosinophil levels and improvements in lung function and exacerbation rates, with greater improvements generally observed with BFF MDI relative to FF MDI in patients with baseline blood eosinophil levels ≥150 cells per mm3.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01334-2018_Supplement
Acknowledgements
The authors would like to thank all of the patients and their families and the team of investigators, research nurses, and operations staff involved in these studies. Medical writing support, under the direction of the authors, was provided by Pauline Craig of CMC CONNECT, a division of Complete Medical Communications Ltd, Glasgow, UK, which was funded by AstraZeneca, Cambridge, UK in accordance with Good Publication Practice (GPP3) guidelines [30].
Footnotes
This article has supplementary material available from erj.ersjournals.com
This study is registered at ClinicalTrials.gov with identifier number NCT02766608. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure
Author contributions: C. Cappelletti, E.A. Duncan, G.T. Ferguson, J. Nyberg and P. Dorinsky made substantial contributions to the conception and design of the work. E.M. Kerwin and G.T. Ferguson made substantial contributions to the acquisition of data. A. Anzueto, A. Papi, C. Cappelletti, E.A. Duncan, E.M. Kerwin, G.T. Ferguson, J. Nyberg and P. Dorinsky made substantial contributions to the analysis of data. A. Anzueto, A. Papi, C. Cappelletti, E.A. Duncan, E.M. Kerwin, G.T. Ferguson, J. Nyberg and P. Dorinsky made substantial contributions to the interpretation of data. All authors were involved in the preparation and review of the manuscript, approved the final version to be submitted and agree to be accountable for all aspects of the work, in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest: G.T. Ferguson reports grants, personal fees and non-financial support from AstraZeneca, during the conduct of the study; and grants, personal fees and non-financial support from Boehringer Ingelheim, Novartis, AstraZeneca, Pearl (a member of the AstraZeneca Group) and Sunovion, grants and personal fees from Theravance, and personal fees from Verona, Mylan, Innoviva, GlaxoSmithKline and Circassia, outside the submitted work.
Conflict of interest: A. Papi reports board membership, consultancy, payment for lectures, grants for research and travel expenses reimbursement from Chiesi, AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Mundipharma and TEVA, payment for lectures and travel expenses reimbursement from Menarini, Novartis and Zambon, board membership, payment for lectures, grants for research and travel expenses reimbursement from Pfizer, and grants for research from Sanofi, outside the submitted work.
Conflict of interest: A. Anzueto reports institutional grants from GSK, and personal fees for consultancy from GSK, AstraZeneca, Novartis and BI, outside the submitted work.
Conflict of interest: E.M. Kerwin is an employee of Crisor LLC Research, has served on advisory boards, speaker panels, or received travel reimbursement from Novartis, AstraZeneca, Amphastar, Forest, Pearl (a member of the AstraZeneca Group), Sunovion, Teva and Theravance, has served on a medical advisory board for Mylan, and has undertaken consultancy for GSK, outside the submitted work.
Conflict of interest: C. Cappelletti is a full-time employee of Pearl (a member of the AstraZeneca Group).
Conflict of interest: E.A. Duncan is a full-time employee of Pearl (a member of the AstraZeneca Group).
Conflict of interest: J. Nyberg is a full-time employee of Pearl (a member of the AstraZeneca Group).
Conflict of interest: P. Dorinsky is a full-time employee of Pearl (a member of the AstraZeneca Group).
This article has supplementary material available from erj.ersjournals.com
Support statement: This study was supported by Pearl – a member of the AstraZeneca Group. The funder of the study was involved in study design, data collection, data analysis, data interpretation and writing of the report. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received July 16, 2018.
- Accepted August 12, 2018.
- Copyright ©ERS 2018
This article is open access and distributed under the terms of the Creative Commons Attribution Non-commercial Licence 4.0.
References










