





Abstract
It is possible that macitentan has therapeutic effects in patients with advanced IPF and associated PH http://ow.ly/NYMs30loVxO
Pulmonary hypertension (PH) is a multi-aetiological haemodynamic and pathophysiological condition defined as an increase in mean pulmonary artery pressure ≥25 mmHg at rest, and classified in five subgroups: 1) pulmonary arterial hypertension (PAH), 2) PH due to left heart disease, 3) PH due to lung disease and/or hypoxia, 4) chronic thromboembolic PH and 5) PH with unclear multifactorial mechanisms [1]. Over the past 20 years, a series of breakthrough therapeutic advances in PAH treatment paved the way for presently four approved drug classes. PAH, however, is a rare disease and the vast majority of PH patients belong to groups 2 and 3 [2], for which no effective therapies have so far been approved [3].
In a study published in this issue of the European Respiratory Journal, Bellaye et al. [4] now report promising results on the therapeutic efficacy of macitentan, an oral dual endothelin-1 (ET-1) receptor antagonist, in a preclinical model of group 3 PH. Macitentan is approved for the treatment of PAH, where it delays disease progression and clinical worsening [5]. Now, Bellaye et al. [4] tested macitentan in a rat model of interstitial pulmonary fibrosis (IPF) with associated PH. Dual endothelin receptor antagonism by bosentan had formerly been shown to attenuate the development of lung fibrosis in the rat bleomycin model [6]. Acute oral administration of both bosentan and macitentan were further shown to reduce pulmonary arterial pressure in the bleomycin rat model; however, only macitentan had a sustained beneficial effect on pulmonary vascular and right ventricular remodelling [7]. These findings point to a potential promise of ET-1 receptor antagonists, and particularly macitentan, in the treatment of IPF and associated PH; yet in both studies ET-1 receptor antagonists were given as prevention rather than treatment, limiting their translational power [8].
Now, Bellaye et al. [4] report elevated circulating levels of ET-1 in IPF patients which correlated with disease severity as assessed by pulmonary function tests. Similarly, expression of the ET-1 receptor A (ETRA) but not of ET-1 receptor B (ETRB) was upregulated in lung tissue of IPF patients as a function of disease stage, suggesting a possible role of ET-1 signalling via ETRA in the disease pathomechanism. Indeed, cumulative evidence points towards a contribution of ET-1 signalling through both ETRA [9] and ETRB [10] to the pathobiology of fibrotic disorders. Specifically, ET-1 has been shown to drive hallmarks of profibrotic cellular responses such as the induction of an enhanced contractile phenotype of fibrotic fibroblasts, their transition to myofibroblasts [11], enhanced collagen production [12], and epithelial-to-mesenchymal transition [13].
Next, Bellaye et al. [4] tested for the effects of macitentan in a rat model of pulmonary fibrosis following adenoviral overexpression of transforming growth factor-β1 (AdTGF-β1) [14]. TGF-β1 is the major cytokine driving both lung fibrosis and PH. Specifically, TGF-β1 induces transition of both epithelial cells [15] and fibroblasts [16] into myofibroblasts, the hallmark cell of IPF. On the vascular side, chronic TGF-β1 stimulation causes smooth muscle proliferation and hypertrophy primarily via a loss of SMAD3 and a subsequent disinhibition of the pro-hypertrophic transcriptional co-factor myocardin-related transcription factor (MRTF) [17]. At the endothelial level, PH is characterised by initial apoptosis and vessel rarefaction followed by subsequent hyperproliferation of apoptosis-resistant cells [18]. In addition, endothelial−mesenchymal transition is increasingly gaining attention as important contributor to vascular remodelling in PH and is likewise proposed to be mediated by TGF-β1 [19], a notion in line with TGF-β1-induced disinhibition of MRTF driving α-smooth muscle actin (α-SMA) expression [17].
Consistent with these established roles of TGF-β1 in lung disease, AdTGF-β1 treated rats developed lung fibrosis and PH. When treated with macitentan between days 14 and 28, the development of PH was significantly attenuated, in that mean pulmonary arterial pressure and vascular wall thickness of pulmonary blood vessels decreased, while density of blood vessels increased in lung histology and lung CT scans. In contrast to macitentan, treatment with pirfenidone, one of two presently approved drugs for the treatment of IPF, had no effect on pulmonary haemodynamics and lung vascular remodelling in AdTGF-β1 treated rats.
In addition to its beneficial therapeutic effects on PH, macitentan also proved effective in preventing the progression of lung fibrosis, in that it reduced collagen deposition and fibrotic area, Ashcroft score, and hydroxyproline content while improving lung function. These therapeutic effects were comparable to those achieved with pirfenidone, with no additive effect when both drugs were combined. The comparison to pirfenidone rules out that positive effects of macitentan on PH in this model were solely attributable to the attenuated progression of lung fibrosis, but instead suggest a specific therapeutic effect of macitentan on group 3 PH.
Additional data by Bellaye et al. [4] suggest at least two potential mechanisms by which macitentan may attenuate the progression of group 3 PH. First, macitentan (but not or to a lesser extent pirfenidone) inhibited endothelial-to-myofibroblast transition, as indicated by prevention of the TGF-β1 upregulation of collagen-1, α-SMA and TGF-β1 in human pulmonary artery endothelial cells (hPAECs). Secondly, macitentan inhibited endothelial apoptosis and vascular rarefaction, i.e. characteristic signs of early vessel loss in PH, both in vitro and in vivo. Specifically, macitentan upregulated vascular endothelial growth factor (VEGF) and downregulated active caspase-3, thereby counteracting the anti-angiogenic and pro-apoptotic effects of TGF-β1 in hPAECs. These in vitro effects were equally evident in vivo as macitentan protected endothelial cells from AdTGF-β1-induced apoptosis while increasing endothelial VEGF expression and counteracting the vascular rarefaction characteristic for PH lungs.
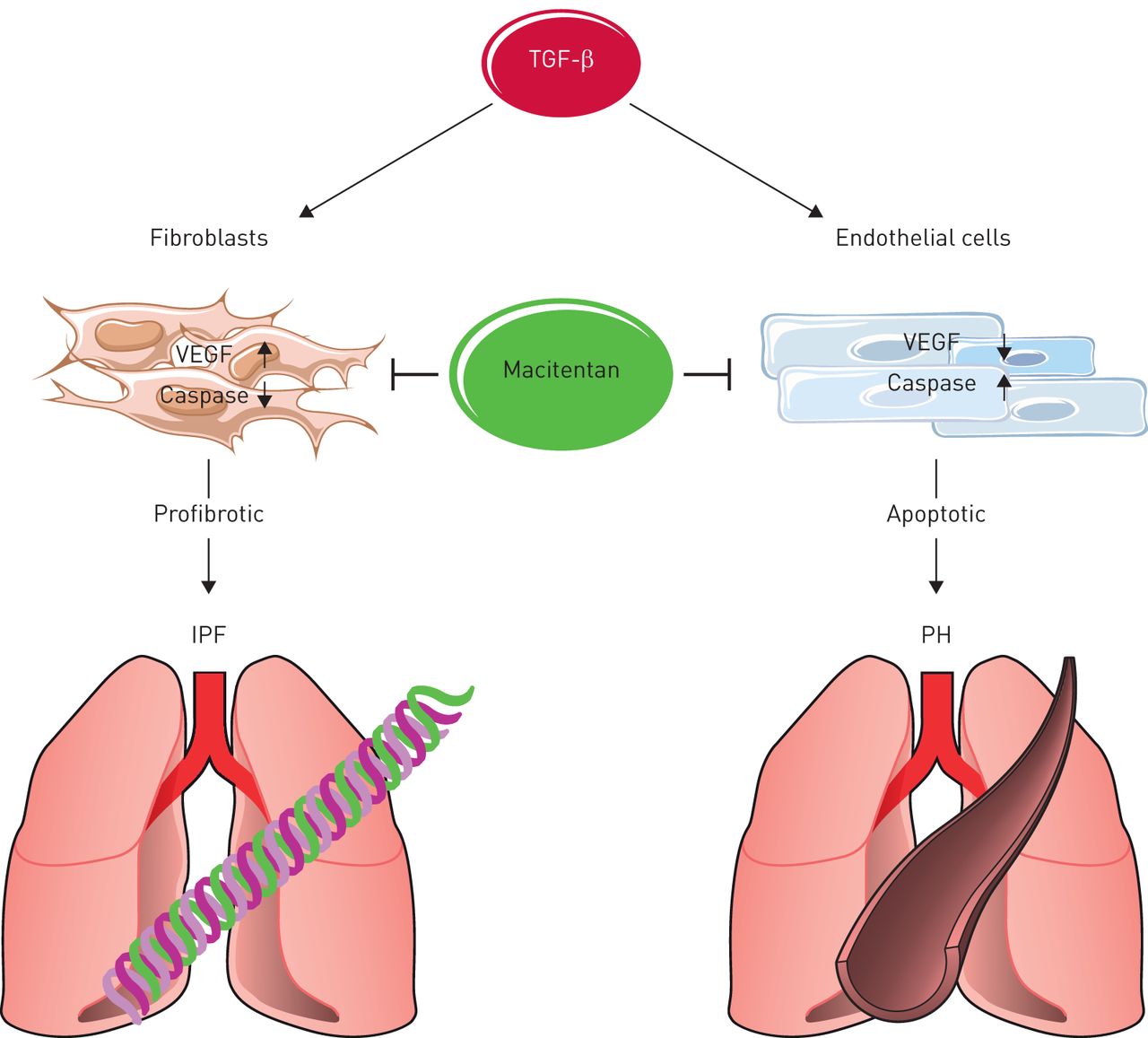
Intriguingly, the latter effects of TGF-β1 and macitentan were diametrically opposed in endothelial cells versus fibroblasts, in that TGF-β1 increased VEGF expression but decreased active caspase-3 in fibroblasts, while VEGF was reduced and caspase-3 activation enhanced in endothelial cells (figure 1). These findings are in line with the notion that TGF-β1 induces both fibrosis progression (via reduced (myo-)fibroblast apoptosis and increased pro-fibrotic VEGF expression in the lung parenchyma) and PH-characteristic vessel loss (via reduced endothelial VEGF abundance and increased endothelial apoptosis). Macitentan prevented both effects, i.e. it attenuated VEGF expression while activating caspase-3 in fibroblasts and lung parenchyma, and concomitantly increased VEGF but prevented caspase-3 activation in endothelial cells. These findings may at least in part explain the dual therapeutic effects of macitentan against both lung fibrosis and PH; yet, the molecular mechanisms underlying the opposing effects of macitentan (and TGF-β1) in endothelial cells versus fibroblasts remain to be elucidated.
{kind=link}
Schematic of the opposing effects of transforming growth factor (TGF)-β1 and its inhibition by macitentan on vascular endothelial growth factor (VEGF) expression and caspase-3 activation in endothelial cells versus fibroblasts driving both TGF-β1-induced lung fibrosis and pulmonary hypertension (PH), and facilitating the dual therapeutic efficacy of macitentan against both lung diseases. IPF: idiopathic pulmonary fibrosis.
With respect to clinical translation, due care and caution are warranted. The previous prospective randomised double-blind, multicentre and placebo-controlled phase II MUSIC trial in 178 IPF patients failed to detect any treatment effects of macitentan over >12 months with respect to pulmonary function tests, time to disease worsening, or death [20], and similar results were reported for the dual ET-1 receptor antagonist bosentan in the BUILD-1 and BUILD-3 trials [21, 22]. However, patients in these trials had mild-to-moderate IPF with no (documented) PH, and it seems conceivable that macitentan's efficacy against IPF may in part result from its therapeutic potency in PH, as the pulmonary vasculature may play a more prominent role in the initiation and/or progression of IPF as commonly considered [23]. For example, strictly endothelial-specific deletion of two gap junctional molecules, connexin 40 and connexin 43, suffices to induce spontaneous lung fibrosis in mice [24]. Similarly, endothelial-specific knockout of the essential autophagy gene Atg7 (autophagy-related protein 7) results in increased susceptibility to bleomycin-induced pulmonary fibrosis and collagen accumulation [25]. In IPF-associated PH, remodelling of pulmonary blood vessels may promote fibroblast activation and fibrosis by providing a stiffer biomechanical context in the lung parenchyma, thereby activating profibrotic mechanosignalling [26, 27]. Bellaye et al. [4] further demonstrate that macitentan prevents TGF-β1 release from hPAECs, which otherwise may amplify fibrosis progression. Taken together, it is possible that macitentan has therapeutic effects in patients with advanced IPF and associated PH that were not evident in the mild-to-moderate patients assessed in the MUSIC and BUILD trials. Notably, while the ARTEMIS trial failed to show a benefit for the ETRA selective receptor antagonist ambrisentan in IPF patients both with and without PH [28], a subsequent analysis of group 3 PH patients showed a reduction in mean pulmonary arterial pressure in ambrisentan-treated patients of 5.3±4.3 mmHg versus 1.1±9.4 mmHg in the placebo arm, and a lower rate of newly developed PH (3% versus 8%); yet overall numbers were small and none of these differences reached significance [29].
Finally, due caution is warranted not to transfer preclinical promise from one group of PH to another. This is a particular concern in group 2 PH (PH due to left heart disease), where at least part of the vascular remodelling is a physiological adaptation to protect the lung from hydrostatic oedema formation in congestive left heart failure [30]. This became again evident in the recent MELODY trial [31], which showed no change in group 2 PH after 12 weeks of macitentan as compared to placebo, but a higher frequency of adverse events and a greater likelihood for fluid retention in line with a higher propensity for oedema formation.
Footnotes
Conflict of interest: J. Grune has nothing to disclose.
Conflict of interest: W.M. Kuebler has nothing to disclose.
- Received July 12, 2018.
- Accepted August 6, 2018.
- Copyright ©ERS 2018
References










