





Abstract
To identify potential risk factors for lung disease progression in children with cystic fibrosis (CF), we studied the longitudinal data of all children with CF (aged ≥5 years) registered in the Dutch CF Registry (2009–2014).
Lung disease progression was expressed as a decline in lung function (forced expiratory volume in 1 s (FEV1) % pred) and pulmonary exacerbation rate. Potential risk factors at baseline included sex, age, best FEV1 % pred, best forced vital capacity % pred, genotype, body mass index z-score, pancreatic insufficiency, medication use (proton pump inhibitors (PPIs), prophylactic antibiotics and inhaled corticosteroids), CF-related diabetes, allergic bronchopulmonary aspergillosis and colonisation with Pseudomonas aeruginosa.
The data of 545 children were analysed. PPI use was associated with both annual decline of FEV1 % pred (p=0.017) and future pulmonary exacerbation rate (p=0.006). Moreover, lower FEV1 % pred at baseline (p=0.007), prophylactic inhaled antibiotic use (p=0.006) and pulmonary exacerbations in the baseline year (p=0.002) were related to pulmonary exacerbations in subsequent years.
In a cohort of Dutch children with CF followed for 5 years, we were able to identify several risk factors for future exacerbations. In particular, the association between PPI use and lung disease progression definitely requires further investigation.
Abstract
The association between the use of proton pump inhibitors and lung disease progression in children with cystic fibrosis requires further investigation http://ow.ly/hiTc30jACAU
Introduction
Respiratory failure is the primary cause of death in patients with cystic fibrosis (CF) [1, 2]. Progression of the lung disease is an ongoing process, aggravated by many factors [3]. In young children, the pulmonary exacerbation rate is an important indicator of lung disease progression [4]. In older subjects, lung disease progression is monitored by the annual decline in forced expiratory volume in 1 s (FEV1). Improving quality of life, prevention of pulmonary exacerbations and preserving lung function are important goals in the management of CF [5]. To prevent pulmonary exacerbations and lung function decline, and thus slow down lung disease progression, it is important to identify risk factors. Some important risk factors for pulmonary exacerbations are known, such as younger age, female sex, use of inhaled corticosteroids (ICSs), level of FEV1, F508del homozygous genotype, low body mass index (BMI), positive respiratory cultures for Pseudomonas aeruginosa and a history of multiple previous exacerbations [6–9].
However, not all risk factors have yet been identified. For instance, more insight into medication use related to disease progression would be interesting. There is an ongoing debate about the safety of proton pump inhibitors (PPIs), which CF patients are frequently prescribed. A recent meta-analysis and systematic review of 26 studies in adults demonstrated that outpatient PPI use was associated with a 1.5-fold increased risk of community-acquired pneumonia (CAP) [10]. In a prospective study in 186 children (without CF) aged 4–36 months, a significant increase in acute gastroenteritis and CAP was also found in patients treated with gastric acid inhibitors compared with healthy controls [11]. As PPI use is common in CF patients, more research on the long-term safety of PPIs in CF is necessary.
From 2008 onwards, the Dutch CF Foundation (www.ncfs.nl) has annually registered data provided by the seven CF centres in the Netherlands. The recorded parameters closely match the European and US patient registries [12]. This national database offers the opportunity to identify risk factors for accelerated annual decline in lung function and for future pulmonary exacerbations in children with CF, and thus to identify children who need closer monitoring. We hypothesised that in conjunction with the more established risk factors (e.g. positive respiratory cultures for P. aeruginosa), the use of medication (PPIs, prophylactic antibiotics, ICSs and pancreatic enzymes), genotype, CF-related diabetes (CFRD) and allergic bronchopulmonary aspergillosis (ABPA) are associated with lung function decline and future pulmonary exacerbations.
Material and methods
Study design
For this longitudinal observational study, the data of patients (5–18 years of age) with CF registered in the Dutch CF Registry from 2009 to 2014 were used. The data were collected during regular patient care. All patients or guardians of young patients provided informed consent for the storage and scientific use of the data. The database of the Dutch CF Foundation is disclosed to the Dutch Data Protection Authority. The diagnosis of CF was confirmed when two known CF-causing mutations were identified (according to the CF transmembrane conductance regulator (CFTR) mutation database CFTR2: www.cftr2.org) and/or a sweat chloride concentration of ≥60 mmol·L−1 was registered [12]. Children <5 years of age at baseline were excluded because lung function data are lacking for this age group.
Study parameters
The first outcome measure was annual change of FEV1 % pred. The best FEV1 % pred of each year (highest value) was recorded according to the definitions of the Global Lung Initiative reference values [13]. The second outcome measure was future pulmonary exacerbations, defined as a course of intravenous antibiotics for pulmonary exacerbation(s). We assessed the effect of possible risk factors at baseline on the chance of having one or more pulmonary exacerbations 1, 2, 3, 4 or 5 years later.
To identify factors that are potentially associated with an annual change of FEV1 % pred and risk of future pulmonary exacerbations, we analysed several variables measured during the first year a patient was registered (baseline). A baseline measure was required to be included in the analysis. This baseline year (based on a calendar year) is not the same for every patient (e.g. patients could enter the study during a calendar year) nor is the duration of follow-up. The baseline variables included sex, age at best FEV1 measurement of the baseline year, best FEV1 % pred and best forced vital capacity (FVC) % pred. In addition, genotype was included and was defined by the number of F508del mutations (homozygote, heterozygote or none; the latter two were combined in the analyses). BMI was also assessed, and z-scores were used to allow for children of different ages and sex to be pooled for the analysis. These scores were calculated with the Growth Analyser application (growthanalyser.org) based on references obtained from the Fifth Dutch Growth Study [14]. Furthermore, we analysed the influence of pancreatic insufficiency, defined by the use of pancreatic enzymes. The influence of PPIs, prophylactic inhaled antibiotics, oral macrolides (as prophylactic antibiotics) and ICSs was also studied. The presence of ABPA was defined by the CF Foundation Consensus Conference in 2003 [15]. CFRD was defined by the use of insulin. Colonisation or chronic infection with P. aeruginosa was defined according to the modified Leeds criteria [16, 17].
Statistical analysis
An extensive description of the statistical analysis is provided in the supplementary material. In summary, for the longitudinal analysis of the first outcome measure (FEV1 % pred), linear mixed effects models were used, in which the numerical variable time (0–5 years), potential risk factors (see earlier) and their interactions with time were entered as fixed factors. As time is entered here as a numerical variable and measured in years, the effect of a potential risk factor on FEV1 % pred indicates whether the annual change in FEV1 % pred is affected by this factor.
For the longitudinal effect on future pulmonary exacerbations (measured at years 1, 2, 3, 4 and 5), generalised estimating equations were used, where time and potential risk factors (including pulmonary exacerbations at baseline) were added as fixed factors. As there is no interaction between time and the potential risk factors, the effect of a potential risk factor is expressed as an overall odds ratio, pooled over all time-points.
There were no restrictions on the minimum follow-up data required for the analysis. Multiple imputation was used to deal with missing data by creating 50 complete datasets. For sensitivity analysis, a complete case analysis was also performed, in which the patients with one or more missing covariate values at baseline were excluded.
For both outcome measures, first univariate (explorative) models and thereafter multivariate models were assessed. The estimated effect (pooled for 5 years) of each potential risk factor on mean annual change in FEV1 % pred or overall odds ratio for pulmonary exacerbations (over all time-points) was reported together with the corresponding 95% confidence interval and p-value. Multicollinearity of the models was checked (all variance inflation factors were <1.7). As a post hoc analysis, we also assessed the association between percentage of years of PPI use and lung disease progression to check the dose–response relationship.
Two-sided p-values ≤0.05 were considered statistically significant. Baseline characteristics and generalised estimating equation analyses were obtained using SPSS Statistics for Windows version 23.0 (IBM, Armonk, NY, USA). The multiple imputation part was performed using the MICE package in R version 3.2.3 [18].
Results
Study participants
Between 2009 and 2014, a total of 860 children with CF (0–18 years) were registered for ≥1 years. Data of 545 children (aged 5–18 years) were analysed. Of those children, 32 (6%) were only registered during 1 year (“baseline”) and 312 (57%) had a maximum follow-up of 5 years. Characteristics of the study cohort in the baseline year are provided in table 1. In 93% of the children, one or two F508del mutations were present, so only 7% might belong to another mutation class. Supplementary table S1 provides a comparison of the baseline characteristics of the CF patients with PPI use in the baseline year versus the children without PPI use.
Baseline characteristics
Risk factors for lung function decline
The mean of best performed FEV1 % pred was 88.5% at baseline, the mean annual decline of FEV1 % pred of the total cohort was −1.92% per year (95% CI −2.26– −1.58% per year).
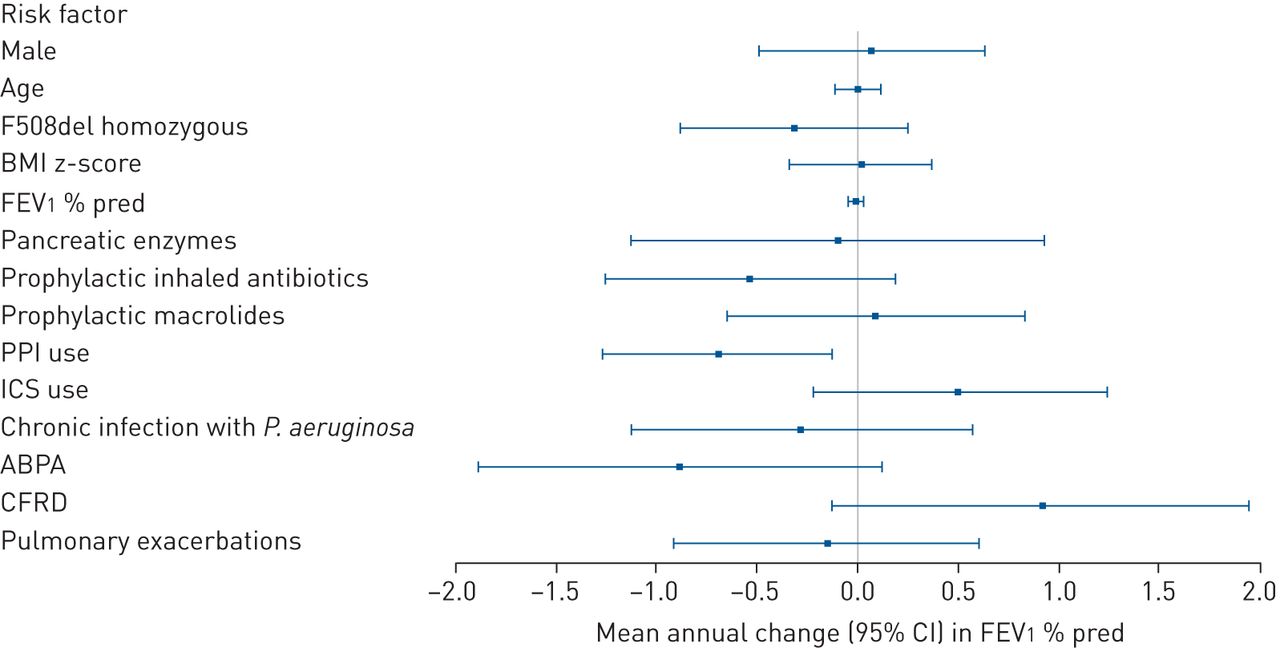
In the univariate analyses, prophylactic inhaled antibiotic use and PPI use were significantly associated with lung function decline (data not shown). Figure 1 and supplementary table S2 show the estimated pooled effect of 5 years of each potential risk factor on mean annual change in FEV1 % pred when applied in a multivariate model. PPI use remained a significant risk factor for a subsequent accelerated annual decline in FEV1 % pred (estimated pooled effect −0.69, 95% CI −1.26– −0.12), after adjusting for all other included risk factors. No other potential risk factors studied had a significant effect on the annual change in FEV1 % pred in this study population. The effects of ABPA and CFRD were of borderline significance (p=0.085 and 0.086, respectively).

Forest plot of adjusted effect of risk factors (measured at baseline) on mean annual change in forced expiratory volume in 1 s (FEV1) % pred. BMI: body mass index; PPI: proton pump inhibitor; ICS: inhaled corticosteroid; P. aeruginosa: Pseudomonas aeruginosa; ABPA: allergic bronchopulmonary aspergillosis; CFRD: cystic fibrosis-related diabetes. The effects were adjusted for the other variables presented here.
Risk factors for pulmonary exacerbations
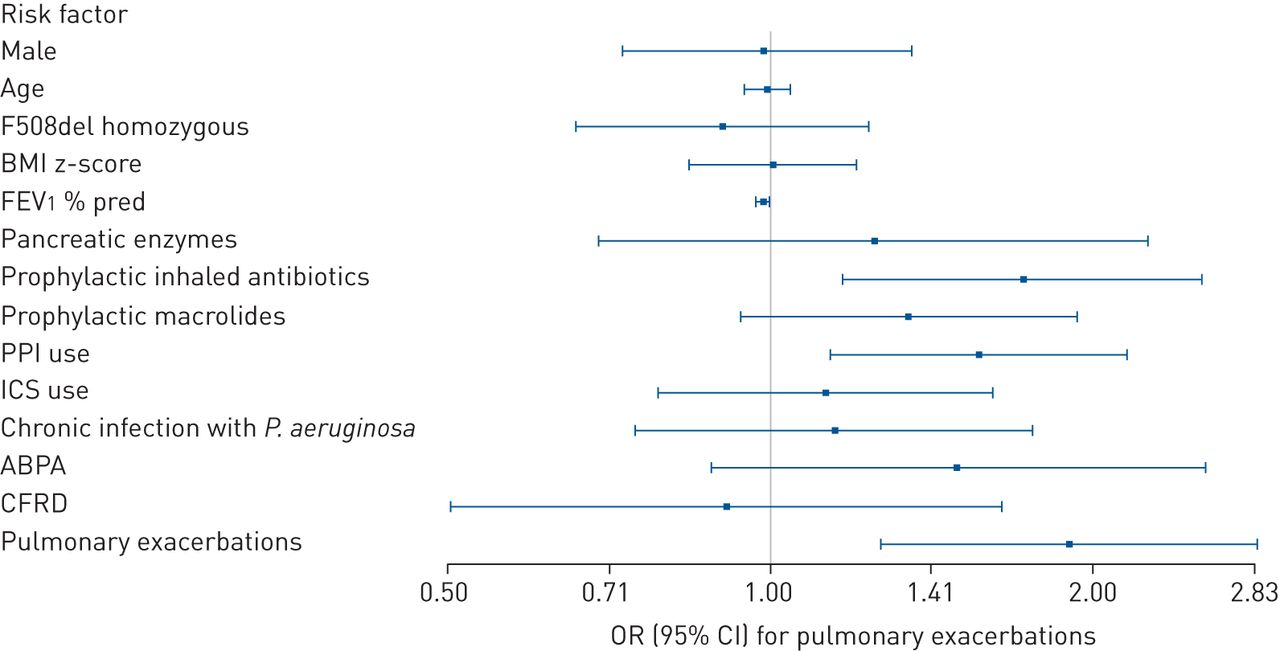
In the univariate analyses, most factors (including colonisation with P. aeruginosa) were significantly associated with pulmonary exacerbations (data not shown). Figure 2 and supplementary table S3 show the odds ratio (pooled for 5 years) for each potential risk factor for future pulmonary exacerbations after correction for the other factors in a multivariate model. A lower FEV1 % pred (OR 0.985, 95% CI 0.974–0.996), prophylactic inhaled antibiotic use (OR 1.722, 95% CI 1.170–2.533), PPI use (OR 1.565, 95% CI 1.138–2.151) and pulmonary exacerbations in the baseline year (OR 1.901, 95% CI 1.272–2.841) were significant risk factors for pulmonary exacerbations in subsequent years (after adjusting for other potential risk factors). In contrast, sex, age, genotype, BMI z-score, colonisation with P. aeruginosa, ABPA, CFRD and the use of pancreatic enzymes or prophylactic macrolides had no significant association with the risk of future pulmonary exacerbations in the multivariate model.

{kind=link}
{kind=link}
Forest plot of adjusted odds ratios (pooled for 5 years) for future pulmonary exacerbations per risk factor (measured at baseline). BMI: body mass index; FEV1: forced expiratory volume in 1 s; PPI: proton pump inhibitor; ICS: inhaled corticosteroid; P. aeruginosa: Pseudomonas aeruginosa; ABPA: allergic bronchopulmonary aspergillosis; CFRD: cystic fibrosis-related diabetes. The effects were adjusted for the other variables presented here.
Association between percentage of years of PPI use and lung disease progression
To examine our finding on the association between PPI use and lung disease progression in more depth, we studied whether the effect of PPI use increased with increasing duration of PPI use (dose–response relationship). These post hoc analyses showed significant relationships between duration of PPI use (the percentage of years PPI was used, e.g. 2 out of 6 years equals 33.3%) and annual decline in FEV1 % pred (p=0.020) and exacerbation rate (p=0.034).
If the percentage of years of PPI use increased by 10%, the mean FEV1 % pred decreased significantly faster over time (estimated pooled effect −0.08, 95% CI −0.16– −0.01; p=0.020). The risk of future pulmonary exacerbations increased significantly with increasing percentage of years of PPI use (pooled OR for 10% increase of PPI use 1.04, 95% CI 1.003–1.08; p=0.034).
Sensitivity analyses
A total of 278 children were included in the complete case analyses for annual decline of FEV1 % pred and future pulmonary exacerbations. Generally, children were excluded because of missing pulmonary exacerbations data at baseline. The baseline characteristics of children included in the complete case analyses were comparable to those who were not included, except for higher percentages of CFRD, antibiotics and ICS use, and a lower BMI z-score in the complete case population (all p<0.05). For both outcomes (annual change in FEV1 % pred and risk of future pulmonary exacerbations), the complete case analyses gave fewer significant results because of wider confidence intervals, although the associations generally follow the same trend (supplementary tables S4 and S5). Only FEV1 % pred (OR 0.983, 95% CI 0.969–0.997; p=0.017) and pulmonary exacerbations at baseline (OR 2.664, 95% CI 1.681–4.219; p<0.001) remained significant for risk of future pulmonary exacerbations. Moreover, the results of sensitivity analyses for the longitudinal effect on pulmonary exacerbations (robust versus model-based estimation; unstructured versus compound symmetric covariance structure) were comparable to the those reported in figures 1 and 2.
Discussion
In this cohort of Dutch children with CF, PPI use was a significant risk factor for both future exacerbations as well as an accelerated annual decline in FEV1 % pred. Moreover, the risk of future exacerbations was significantly elevated in children with lower FEV1 % pred, those using prophylactic inhaled antibiotics and those with a higher exacerbation rate in the past.
Although there is good evidence for a relationship between PPI use and an increased risk of respiratory tract infections including CAP in non-CF populations [10, 19], there are hardly any data regarding CF populations. To the best of our knowledge, our study is the first longitudinal, multicentre study on CF showing that PPI use was significantly associated with both future pulmonary exacerbations and the annual decline in FEV1 % pred. In CF, a PPI is often added to pharmacological treatment in order to improve gastrointestinal symptoms, fat absorption and thereby nutritional status, as recently reviewed [20]. How can we explain the relationship between PPI use and the lung disease progression of CF in our study?
First, PPI use has been related to proton pumps located in the upper and lower respiratory tract, suggesting that pH dysregulation may alter the respiratory flora and microbiome in the airways, thereby directly potentiating respiratory pathogens and infection [10]. A systematic review of 33 studies with 6 351 656 participants (without CF) showed an increased risk of pneumonia among PPI users compared with subjects not taking PPIs. The risk was highest in the first month after initiation of therapy [10]. One explanation may be that pathogenic bacteria are normally killed by the acid conditions in the stomach and that this mechanism is negatively influenced by using PPIs. As a consequence and with gastro-oesophageal reflux episodes, these pathogenic bacteria can reach the larynx/glottis and can be aspirated into the airways. We expect that this mechanism could also be of importance in CF patients. In a small randomised controlled trial (RCT) in 17 adults with CF, treatment with esomeprazole 40 mg twice daily versus placebo resulted in a trend to earlier and more frequent exacerbations in the esomeprazole group [21]. However, our observational findings should be interpreted with care. It is important to conduct an RCT in a larger CF population and to assess possible changes in the microbiome in the upper and lower airways in support of this hypothesis.
Second, it should be ruled out whether PPI use or gastro-oesophageal reflux disease (GORD) itself is the reason for lung disease deterioration. Both can lead to altered gastric colonisation with increased risk of aspiration of pathogenic bacteria. In a retrospective cohort study, it was demonstrated that children with CF who received gastric acid inhibition for GORD had reduced pulmonary function and earlier acquisition of P. aeruginosa and Staphylococcus aureus at follow-up. In contrast, gastric acid inhibition use in children taking the medication for fat malabsorption (instead of GORD) did not exhibit any negative influence, pointing to a central role of GORD [22]. In line with this, Reen et al. [23] demonstrated a marked reduction in biodiversity of the microbiome in the sputum of CF patients who were symptomatic for GORD compared with those who were not. Unfortunately, no information on PPIs was given. However, the data suggest that GORD and bile aspiration may play a role in airway disease in CF, negatively impacting the microbiome.
Third, the relationship between PPI and lung disease progression in CF may be confounded by disease severity and poor nutritional status, for example. GORD is more frequent in CF patients and is present in almost all patients with end-stage lung disease [24]. Therefore, the use of PPIs might be a proxy for disease severity. However, we think there are several reasons that make reverse causation less likely. Most importantly, we found significant relationships between the duration of PPI use and the annual decline in FEV1 % pred and exacerbation rate. We found no clinical or statistical significant differences in age, sex, lung function, use of prophylactic antibiotics and presence of ABPA between those using PPIs and those not using PPIs. The observed difference in use of pancreatic enzymes (and pancreas insufficiency) between these groups could be expected as this is clearly linked to the indication for prescribing PPIs, which may also explain the small clinical difference in BMI z-score between the groups (difference of −0.18). ICS use and CFRD were not related to the outcome measures (pulmonary exacerbations and decline in FEV1) in our study and therefore cannot explain or bias the results of our study. We corrected for potential confounding factors including those related to disease severity (e.g. lung function, genotype, Pseudomonas colonisation and prior pulmonary exacerbations). We previously demonstrated that CF enteropathy was related to PPI use [25]. Two recent studies found that a low BMI was associated with an increased decline in lung function [8, 9]. However, our present findings demonstrate that after correction for factors reflecting disease severity (e.g. BMI z-score, baseline FEV1 % pred and pulmonary exacerbations), PPI use was still a significant risk factor for lung disease progression.
Taken together, prospective RCTs are needed to shed more light on the relationship between PPI use and lung disease progression in CF.
In addition to PPI use, we found that the risk for future pulmonary exacerbations was significantly higher in patients with a lower FEV1 % pred, which is in keeping with previous studies [7]. Lung function decline can be the consequence of increased airway obstruction, restrictive changes or irreversible alterations such as fibrosis and bronchiectasis [26]. These changes create an environment in which various microbes can more easily colonise the airways, resulting in episodic rapid increases in numbers associated with respiratory infections and pulmonary exacerbations in CF lungs [27].
The use of prophylactic inhaled antibiotics was also associated with a higher odds ratio for future exacerbations. A side-effect of the frequent use of antibiotics could be a decrease in the diversity of the normal airway microbiome, with an increased risk of selection of more pathogenic bacteria [28], such as P. aeruginosa. In a univariate model, Pseudomonas colonisation became a significant risk factor for future pulmonary exacerbations. This last finding coincides with multiple previous findings [8, 29–31].
In our analysis, pulmonary exacerbations in the past was a risk factor for future exacerbations. This indicates that specific patients are at greater risk of experiencing exacerbations, although the reason is unclear. We speculate that the presence of bronchiectasis [32], colonisation with microbial pathogens and/or patient factors, such as reduced use of bronchial clearance techniques and lower adherence to medication, may be responsible for this effect. In chronic obstructive pulmonary disease and asthma, some patients are known to consistently have more exacerbations than others (referred to as “frequent exacerbator phenotype”) [33, 34].
A strength of our study is the longitudinal, multicentre study design with data collection over the course of several years. The missing values for pulmonary exacerbations and the need for multiple imputation to deal with these missing values comprise a limitation of our study. To increase insight regarding this limitation, we performed a complete case analysis. We found fewer significant relationships, which are probably due to the loss in power. Another limitation is that we have no information regarding pulmonary exacerbations treated with oral antibiotics, colonisation with other bacteria or socioeconomic status, which could also be potential risk factors for lung disease progression. Finally, the observational design does not allow us to draw conclusions about causality.
The lack of effects of genotype, use of pancreatic enzymes, ABPA and CFRD in this study should be interpreted with care due to the small number of children without a F508del mutation (future studies should focus on class definitions), not using pancreatic enzymes and with a diagnosis of ABPA or CFRD, respectively. In contrast to other studies, we did not find an effect of nutritional status on disease progression, probably because of the relatively good nutritional status of this cohort (demonstrated by only slightly negative BMI z-scores) [8, 35, 36]. Further studies are needed to elucidate the exact effect of nutritional status on lung disease progression (e.g. including other measures such as “weight for age”). Future research should also take CFTR potentiators/correctors into account in their analysis.
In this longitudinal study in 545 children with CF (≥5 years of age), PPI use was associated with an unfavourable course of the disease (as reflected by both an accelerated decline in FEV1 % pred and increased exacerbation rate). The risk regarding future pulmonary exacerbations was increased in patients with a lower FEV1 % pred, use of prophylactic antibiotics and exacerbations in the past. The potential risk associated with PPI use in particular deserves further investigation.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02509-2017_Supplement
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: None declared.
Support statement: The national database used in this study is funded, coordinated and hosted by the Dutch CF Foundation, and was represented by one of the co-authors (V. Gulmans) who assisted in the interpretation of the data and writing of the report. The corresponding author had full access to all of the data in order to analyse and interpret the data, and had the final responsibility for the decision to submit for publication. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 20, 2017.
- Accepted April 16, 2018.
- Copyright ©ERS 2018
References










