





Abstract
Genetic defects in bone morphogenetic protein type II receptor (BMPRII) signalling and inflammation contribute to the pathogenesis of pulmonary arterial hypertension (PAH). The receptor is activated by bone morphogenetic protein (BMP) ligands, which also enhance BMPR2 transcription. A small-molecule BMP upregulator with selectivity on vascular endothelium would be a desirable therapeutic intervention for PAH.
We assayed compounds identified in the screening of BMP2 upregulators for their ability to increase the expression of inhibitor of DNA binding 1 (Id1), using a dual reporter driven specifically in human embryonic stem cell-derived endothelial cells. These assays identified a novel piperidine, BMP upregulator 1 (BUR1), that increased endothelial Id1 expression with a half-maximal effective concentration of 0.098 μmol·L−1. Microarray analyses and immunoblotting showed that BUR1 induced BMP2 and prostaglandin-endoperoxide synthase 2 (PTGS2) expression. BUR1 effectively rescued deficient angiogenesis in autologous BMPR2+/R899X endothelial cells generated by CRISPR/Cas9 and patient cells.
BUR1 prevented and reversed PAH in monocrotaline rats, and restored BMPRII downstream signalling and modulated the arachidonic acid pathway in the pulmonary arterial endothelium in the Sugen 5416/hypoxia PAH mouse model.
In conclusion, using stem cell technology we have provided a novel small-molecule compound which regulates BMP2 and PTGS2 levels that might be useful for the treatment of PAH.
Abstract
Stem cell technology identified a novel small-molecule compound that regulates BMP2 and PTGS2 levels in PAH http://ow.ly/l8R630iqf1R
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease initiated by endothelial dysfunction and apoptosis. It results in elevated pulmonary arterial pressure and right ventricular failure [1]. It has been proposed that dysfunctional endothelial cells have impaired cell–cell junctions and high permeability, which allows proinflammatory factors to infiltrate the smooth muscle layer and induce proliferation [2]. Although there have been substantial advances in PAH therapeutics, the disease is still associated with high mortality [3, 4]. Current therapies, including endothelin receptor antagonists, phosphodiesterase 5 inhibitors and prostacyclin analogues, mainly affect pulmonary arterial vasoconstriction [5]. Additional approaches are needed that directly improve the vascular remodelling that characterises PAH pathology [6].
Bone morphogenetic protein (BMP) signalling plays a pivotal role in the development of the vascular system [7] and is involved in the maintenance and homeostasis of adult vasculature [8]. Heterozygous mutations in the gene encoding BMP type II receptor (BMPRII), a receptor for the transforming growth factor-β superfamily, have been found in ∼80% of familial PAH cases and in 15–20% of idiopathic PAH cases. Moreover, even in the absence of identifiable mutations, BMPRII expression in the pulmonary vasculature is markedly reduced in idiopathic PAH [9–11]. Together, these findings suggest that rescuing BMPRII signalling with small molecular regulators with low toxicity may be a tractable approach for treating PAH [12].
Maintaining a balance between local production of endothelial-derived prostanoids and leukotrienes is important to maintain homeostasis in the pulmonary vasculature. Prostaglandin-endoperoxide synthase 2 (PTGS2, also known as cyclooxygenase-2) is induced by hypoxia to increase prostaglandin E2 production in endothelial cells. Homozygous PTGS2 knockout mice developed severe PAH under chronic hypoxia. Conversely, arachidonate 5-lipoxygenase activating protein binds arachidonic acid and transfers it to arachidonate 5-lipoxygenase (ALOX5), subsequently catalysing it into leukotrienes like leukotriene B4 (LTB4), high levels of which have been detected in PAH patients and the Sugen 5416/athymic rat model of pulmonary hypertension [13]. Moreover, an ALOX5 inhibitor effectively reduced pulmonary vascular remodelling in chronic Sugen 5416/hypoxia (SU/Hy)-exposed rats [14].
Here, we report a human embryonic stem cell (hESC)-derived endothelial cell-based Id1-Venus-Luciferase dual reporter system and provide a small-molecule BMP upregulator (BUR1) that activates BMPRII signalling in autologous BMPR2+/R899X human endothelial cells and patient cells via BMP2. Microarray assays revealed that PTGS2 was also upregulated after treatment with BUR1. In vivo, BUR1 not only prevented and reversed PAH in monocrotaline (MCT) rats but also had effects in the SU/Hy rat model.
Materials and methods
Chemical compound screening
Conventional H9 hESCs (Wicell, Madison, WI, USA) were stably transfected with a construct (Id1-Venus-Luc-MC1-DTA) in which venus green fluorescent protein (GFP) and firefly luciferase were driven by the full-length human Id1 promoter (±5 kilobytes). The reporter cells were seeded in 96-well plates and 31 candidate chemical compounds were assigned to each well (obtained from the synthetic and natural library of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Science, Beijing, China). After 4 h, the luciferase activity of each well was calculated using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA).
Generation of R899X heterozygous mutant lines
The H9 hESCs were co-transfected with 30 μg of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 plasmids and 30 μg of donor plasmids in E8 medium overnight via Lipofectamine® 3000, following the manufacturer's instructions. GFP-positive cells were sorted and the genomic DNA of colonies was extracted. Sequence primers are listed in supplemental table S1.
SU/Hy rat model
The SU/Hy rat model was established according to the details described previously [15]. At the end of a 6-week model establishment, 4.5 mg·kg−1 of BUR1 or the vehicle control 0.5% carboxymethyl cellulose was administrated daily via the intragastric (i.g.) route for three more weeks before mice underwent cardiopulmonary phenotyping and were killed.
Quantification of PGE2 and LTB4 in the rat plasma
Blood samples were collected from rats in the SU/Hy model assay. Prostaglandin E2 (PGE2) and LTB4 levels in the plasma were measured using the Prostaglandin E2 high sensitivity ELISA Kit (Abcam, Cambridge, UK) and the LTB4 Parameter Assay Kit (R&D Systems, Minneapolis, MN, USA) with a competitive ELISA method according to the manufacturers' instructions.
Additional information about reporter line generation, microarrays, endothelial cell differentiation from hESCs, CRISPR/Cas9-based mutation generation, immunostaining, MCT, SU/Hy animal models and other experiments can be found in the supplemental material.
Results
Design and validation of a reporter hESC cell line for BMPRII signalling
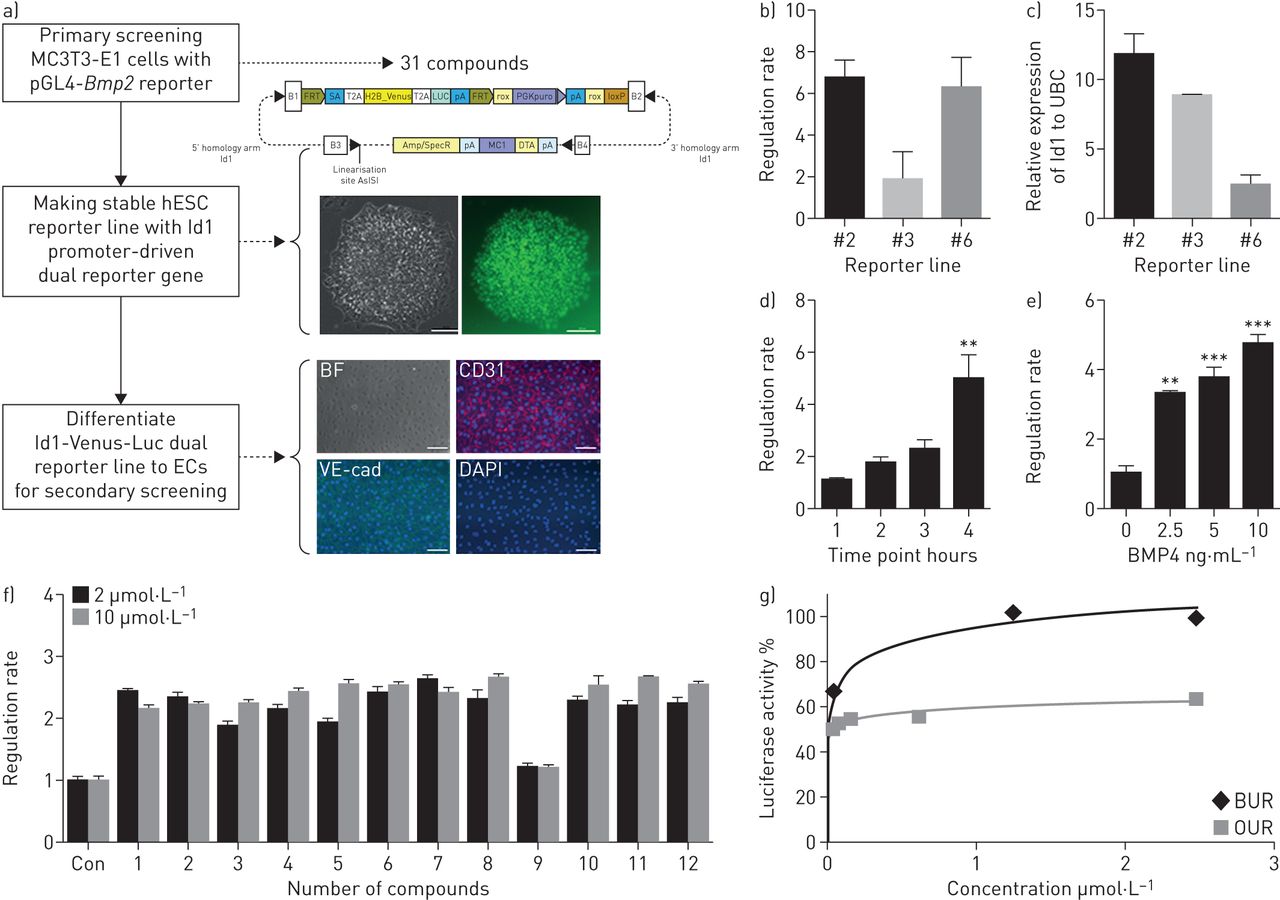
BMPs, including BMP9 and BMP2, increase BMPRII and downstream signalling in vascular cells [15, 16]. A small chemical compound modulating BMP9 or BMP2 needs to be identified for clinical application. Using BMP2-LUC-MC3T3 stable cells, 31 compounds were identified from a previous screening of 8160 small molecular chemicals for their ability to regulate BMP2 expression [17]. These 31 compounds comprised 12 BURs and 19 osteoprotegerin (OPG) upregulators (OURs). To test the ability of these compounds to activate BMPRII signalling on endothelial cells, we generated hESC-Id1-Venus-Luc dual reporter lines with a luciferase gene driven by the full-length Id1 promoter for further validation (figure 1a). Although clone 2 (CGMCC11091) did not show the highest activity upon BMP4 stimulation [18, 19], this clone was selected for validation because the luciferase activity in this cell line correlated well with the induced Id1 mRNA level in the presence of BMP4. This clone's luciferase activity induced by BMP4 demonstrated a time- and concentration-dependent pattern (figure 1b). As demonstrated by the Z'-factor (0.76), the response of this cell line was considered suitable for a high-throughput screening assay (supplemental table S2) [17]. Among the 31 candidate compounds, most of the 12 BURs displayed more than 2-fold luciferase activity compared with vehicle control, and reached a high activity plateau at a lower dose than did the 19 OURs (figure 1c, d).

Stem cell-based screening for activators of BMPRII signalling. a) Schematic outline of the primary screening for BMP2 upregulators and generation of dual reporter human embryonic stem cell (hESC)-derived endothelial cells (ECs) for secondary screening of activators of Id1 transcription. See the text for further details. Representative bright field (BF) and fluorescent images of the generated Id1-Venus-Luc dual reporter hESCs (H9). The Id1-Venus-Luc dual reporter line was differentiated into ECs; fluorescent images show CD31, VE-cadherin (VE-cad) and DAPI-stained nuclei. Scale bars, 100 μm in both BF and fluorescent images. b) Id1-Venus-Luc activity was detected in three different stable lines after 4 h of treatment with or without 10 ng·mL−1 BMP4. c) Id1 mRNA normalised to ubiquitin C (UBC) mRNA in the same three reporter lines (n=3). d) Id1-Venus-Luc activity was determined at different time points (with or without 10 ng·mL−1 BMP4, 1–4 h) (n=4) or e) with different concentrations of BMP4 (0, 2.5, 5, 10 ng·mL−1) after 4 h of treatment (n=4). f) Cells were incubated for 24 h with 0.1% DMSO (control (Con)) or 2 μg·mL−1 or 10 μg·mL−1 of compounds from 31 candidate chemicals; the relative luminescence units were then detected. Graph displays representative examples of the regulation rate by most BUR series compounds (more than 2-fold increase over control) (n=3). f) Percentage luciferase activity of representative BUR1 and OUR4 compounds (n=3). **p<0.01, ***p<0.001.
Identification of a novel BMP2 upregulator in human endothelial cells
To further validate the effectiveness of the BUR series of compounds on vascular cells, we differentiated CGMCC11091 into CD31-positive cells [20] (figure 1a and supplemental methods). BUR1 significantly increased the Id1 promoter-driven luciferase activity at the nanomolar level in CD31-positive endothelial cells. BUR1 (1-{1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl}piperidine) was more effective than the other 11 BUR compounds, showing the lowest half-maximal effective concentration (EC50) at 0.098 μmol·L−1. Moreover, BMPRII was upregulated by BUR1 in human pulmonary arterial endothelial cells (PAECs). Thus, it was selected for further analysis (figure 2a–c). The maximum regulation rate (luciferase activity/control luciferase activity) achieved using 1 μmol·L−1 of BUR1 was 2.23. To understand the activity of BUR1 in endothelial cells, we used BUR1 to stimulate blood outgrowth endothelial cells (BOECs) from a PAH patient for 24 h as a secondary screening and assessed the transcriptomic changes using microarray analysis. BUR1 induced the differential regulation of 112 genes (adjusted p-value 0.05), including BMP2 and PTGS2. Although other BMP ligand transcripts appeared when cells were stimulated with a low dose of BUR1, BMP2 was consistently upregulated in all tests performed (figure 2d). The enhanced expression of BMP2 and PTGS2 was confirmed by quantitative real-time PCR after 24 h BUR1 stimulation (supplemental figure S1a). Quantitative real-time PCR and immunoblotting also demonstrated that expression of BMP2 and PTGS2 had increased after 4 h of BUR1 stimulation (figure 2e, f).

Identification of a novel BMP2 upregulator in human endothelial cells. a) Luciferase activity of Id1-Venus-Luc human embryonic stem cell (hESC)-derived endothelial cells (ECs) upon stimulation with serially diluted concentrations of BUR1 (0.05–1 μmol·L−1). b) Chemical structure of BUR1. c) Representative immunoblotting shows the protein expression levels of BMPRII following stimulation with BUR1 at the indicated concentrations in pulmonary arterial endothelial cells (PAECs). The results are normalised against GAPDH (n=3). d) Volcano plots of differentially expressed genes in blood outgrowth endothelial cells treated with 1 μmol·L−1 BUR1 (n=3, p<0.05, one-way ANOVA). e) Relative mRNA expression level of BMP2 in PAECs after stimulation with 0.1, 1, 10 or 25 µmol·L−1 BUR1 in medium containing 0.1% fetal bovine serum (FBS) for 4 h; results are normalised to GAPDH (n=3). Representative immunoblotting shows the protein expression levels of BMP2 following stimulation with BUR1 for 4 h at the indicated low concentrations in PAECs. f) Relative mRNA expression level of PTGS2 in PAECs after stimulation with BUR1 at 0.1, 1, 10 or 25 µmol·L−1 in medium containing 0.1% FBS for 4 h (n=3). Representative immunoblotting shows the protein expression levels of PTGS2 following stimulation with BUR1 for 4 h at the indicated concentrations in PAECs. The PAECs of passages 4 and 6 were applied in these experiments. **p<0.01, ***p<0.001, ****p<0.0001. EC50: half-maximal effective concentration; Con: control; TGF-β; transforming growth factor β; VEGF: vascular endothelial growth factor.
Low doses of BUR1 enhance the expression of Id1 and phosphorylation of Smad1/5 via BMPRII and ActRII in PAECs
We treated cells with BUR1 for various durations from 15 min to 24 h. Smad1/5 phosphorylation peaked at 30 min, with maximum stimulation and nuclear translocation occurring at 1 h; BUR1 rapidly induced Id1 protein at 15 min, reached a plateau at ∼1 h and showed a prolonged increase in expression to 24 h (figure 3a, supplemental figure S1). To evaluate the specificity and effectiveness of BUR1 in PAECs, we treated cells with low doses of BUR1 at 1 h; BUR1 induced Id1 expression in PAECs in a dose-dependent manner (figure 3b). At a concentration of 0.2 μmol·L−1, BUR1 enhanced Id1 and Id2 mRNA expression in PAECs (∼2.2-fold). A nanomolar level of BUR1 increased BMPR2 mRNA, which confirmed the regulation of BMPRII signalling by BUR1 at the transcriptional level (figure 3c–e, supplemental figure S1a). Considering the consistency of transcriptional and translational regulation of BMPRII downstream effectors, a concentration of 0.2 μmol·L−1 was used for the following experiments.

Low doses of BUR1 enhance canonical BMP signalling via BMPRII and ActRII. a) Representative immunoblotting and relative densitometric analyses of pulmonary arterial endothelial cells (PAECs) show the protein expression levels of p-Smad1/5 and Id1 following stimulation with either 0.2 μmol·L−1 BUR1 at different time points or 1 ng·mL−1 BMP9 for 1 h; the results are normalised to GAPDH (n=3). b) Representative immunoblotting and relative densitometric analyses of PAECs show the protein expression levels of Id1 and p-Smad1/5 following stimulation with either a low dose of BUR1 (0.01, 0.05, 0.1 or 0.2 μmol·L−1) or 1 ng·mL−1 BMP9; the results are normalised to GAPDH (n=3). c) Relative mRNA expression levels of BMPR2 detected in the unstimulated (control (Con)) or stimulated PAECs after treatment with low doses of BUR1 (0.1, 0.2 or 0.5 μmol·L−1) for 1 h. BMP9 (10 ng·mL−1) was used as a positive control; the results are normalised to GAPDH (n=3). d, e) Relative mRNA expression levels of Id1 (d) and Id2 (e) had the same detection patterns after treatment with low doses of BUR1 (0.01, 0.05, 0.1 or 0.2 μmol·L−1) (n=3). f) Representative immunoblotting and relative densitometric analyses of PAECs show the protein expression levels of p-Smad1/5 and Id1 following stimulation with 0.2 μmol·L−1 BUR1 for 1 h, with or without pre-incubation with different concentrations (0.01–1 μmol·L−1) of the kinase inhibitor LDN-193189 for 30 min; the results are normalised to GAPDH (n=3). g) Immunoblotting with monoclonal antibody shows the effects of BMPRII (BRII)- and/or ActRII (ARII)-specific siRNA on Id1 protein expression; the densitometry results are shown below (n=3). h) Immunoblotting with the Id1 antibody shows the effects of individual siRNAs (type I receptor) on Id1 protein expression. The results are normalised to GAPDH (n=3). The PAECs of passages 4 and 6 were applied in these experiments. FBS: fetal bovine serum: NC: non-targeting control siRNA. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To further investigate which receptors mediate the activation of BMPRII signalling, we employed the selective inhibitor LDN-193189 to block the BMP type I receptor-mediated pathway. LDN-193189 attenuated downstream signalling in a dose-dependent manner (figure 3f). When we used BMP9 in place of serum, inhibition by LDN-193189 was still observed (supplemental figure S2). Next, we confirmed that BUR1 and BMP9 had a synergistic effect on the phosphorylation of Smad1/5 (supplemental figure S3). Only co-treatment with activin A receptor type II (ActRII) and BMPRII siRNA reduced the Id1 expression to basal levels (figure 3g). Activin A receptor type 1 (ALK2) knockdown also reduced BUR1-mediated phosphorylation of Smad1/5 (figure 3h and supplemental figure S4). A truncated BMP2 promoter assay demonstrated that the Smad binding element, not the retinoic acid responsive element, was responsible for BMP2 transcription activation by BUR1 stimulation (supplemental figure S5). We also tested other cell types, including BOECs, human umbilical vein endothelial cells, cardiomyocytes and human pulmonary arterial smooth muscle cells. The expression of BMPRII was increased by BUR1 in BOECs and human umbilical vein endothelial cells, but not in the cardiomyocytes. Pulmonary arterial smooth muscle cells showed mild increases in BMP2 protein expression, as did cardiomyocytes, and a smaller change in BMPRII signalling compared to endothelial cells. PTGS2 was also elevated by BUR1 treatment in the cardiomyocytes (supplemental figure S6). Taken together, these findings suggested that BUR1 upregulates BMP2 through the Smad binding element, then enhances BMPRII signalling via ALK2 mainly in endothelial cells.
BUR1 enhances canonical and non-canonical BMP signalling and reverses tube formation defects in BMPRII-deficient endothelial cells
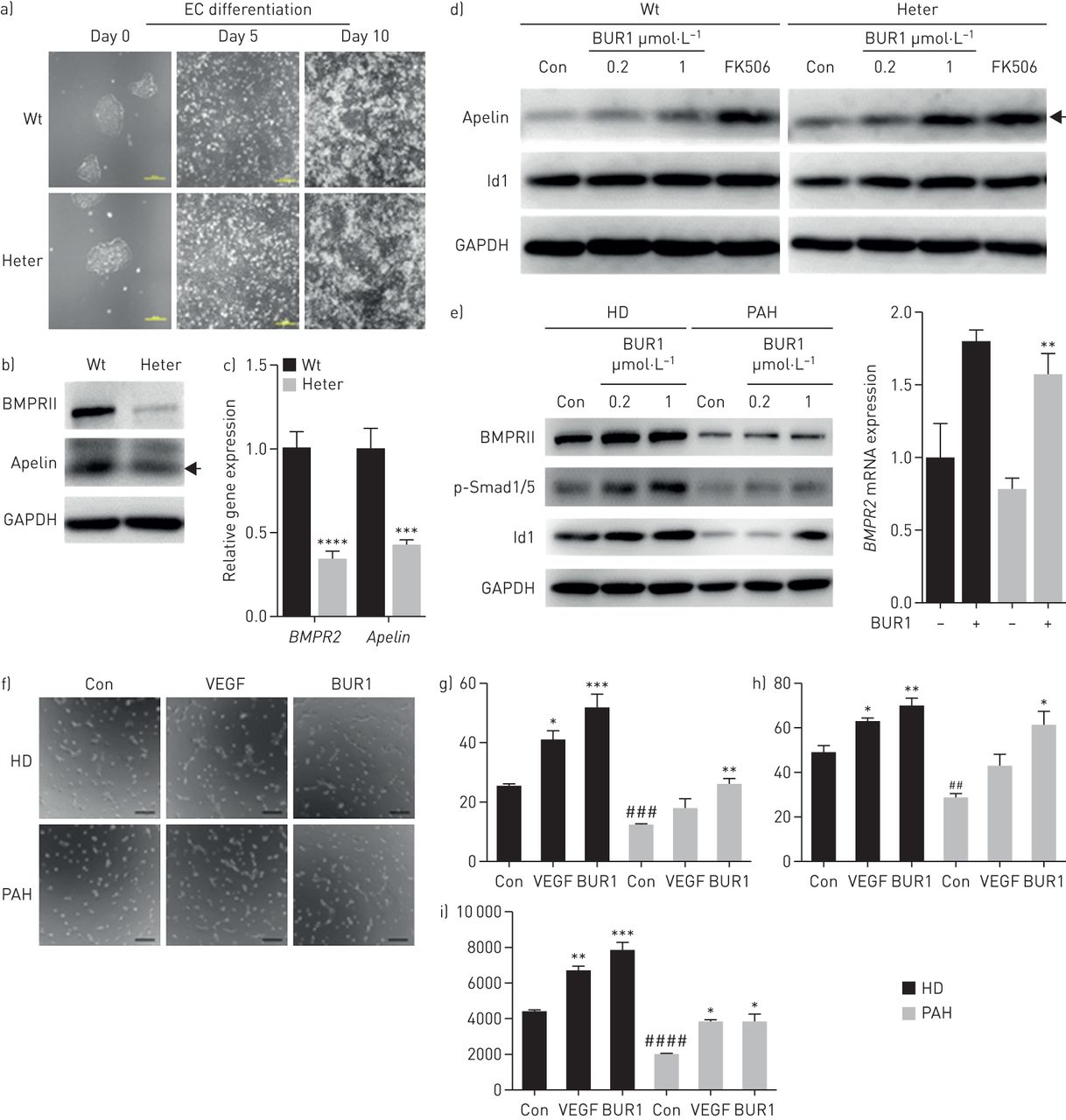
To test the effects of BUR1 on heritable PAH (HPAH) caused by a premature stop mutation, we employed the CRISPR/Cas9 system to generate the R899X autologous heterozygous mutation (figure 4a–d, supplemental figure S7, supplemental tables S3 and S4). To assess the effects of haploinsufficiency on vascular formation, we differentiated the targeted lines in parallel with autologous wild-type hESCs. The expression of Apelin, a ligand that specifically binds the G protein-coupled receptor APJ in endothelial cells [21], was significantly reduced in the heterozygous cell line (figure 4a–c) [22]. We employed differentiated BMPR2+/R899X endothelial cells to assess BMPRII signalling in response to BUR1, and we observed enhanced Apelin and Id1 expression (figure 4d). Using BOECs from patients with idiopathic PAH, we also demonstrated that BUR1 enhanced BMPRII signalling (figure 4e). Furthermore, BUR1 rescued tube network formation similarly to vascular endothelial growth factor in patient endothelial cells (figure 4f–i) and BMPR2+/R899X endothelial cells (supplemental figure S7d). Although Id1 is known to enhance the stability of hypoxia-inducible factor (HIF) proteins to promote tube formation, we found that HIF-1α, HIF-2α and its downstream target arginase-1 were not significantly affected in cultured PAECs or in the pulmonary arteries of SU/Hy-treated rats (supplemental figure S8).
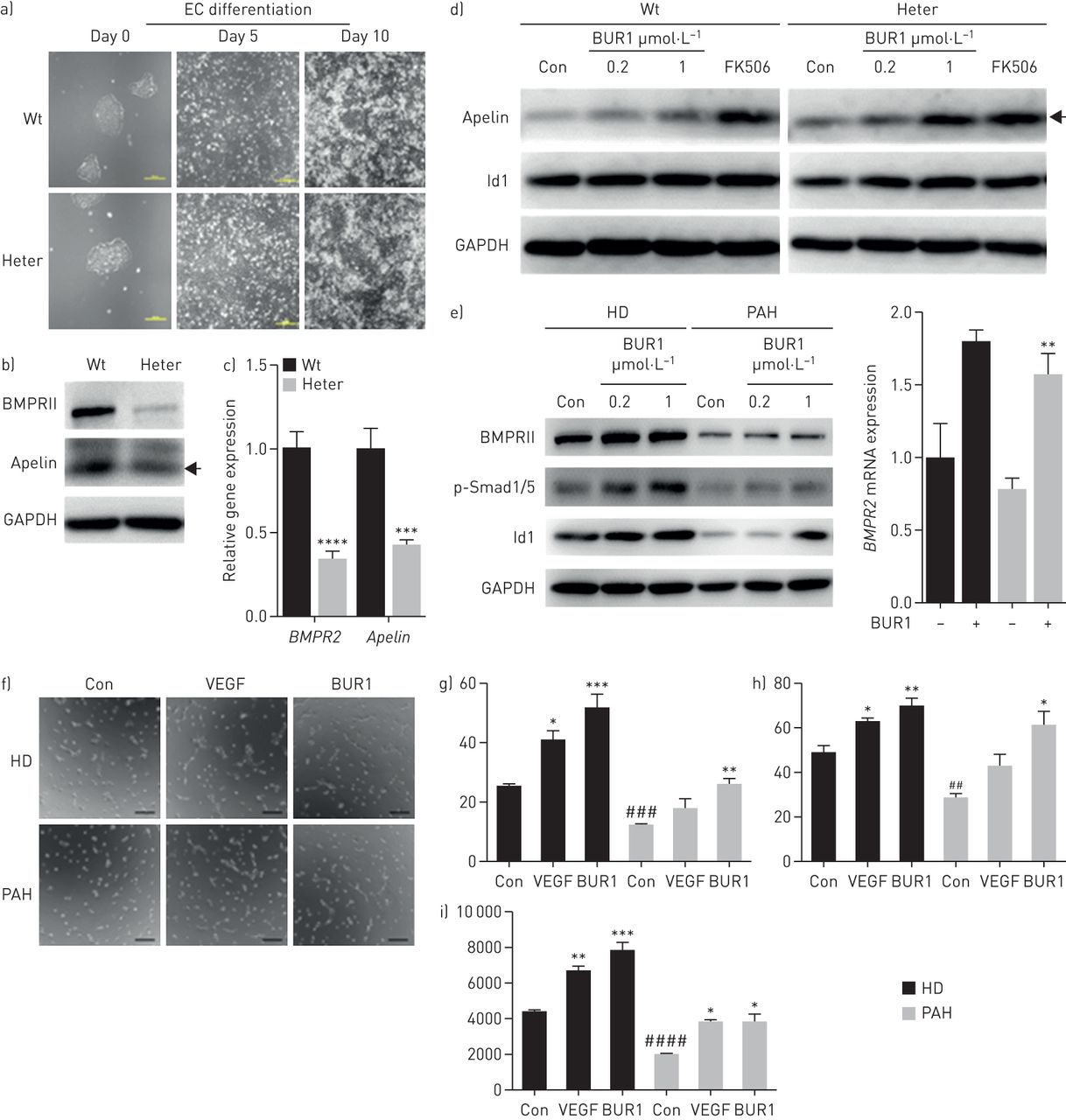
BUR1 enhances BMP signalling and tube formation in autologous BMPR2+/R899X endothelial cells (ECs) and blood outgrowth endothelial cells (BOECs) from a patient with pulmonary arterial hypertension (PAH). a) Morphological changes and relative mRNA expression of BMPR2 upon EC induction at day 0, day 5 and day 10 in BMPR2 wild-type (Wt) and heterozygous (BMPR2+/R899X, Heter) cell lines. Scale bars, 200 μm. b) Immunoblotting and c) quantitative real-time PCR results show the protein levels and mRNA levels of BMPRII and Apelin in Wt and Heter ECs, respectively (n=3). d) Protein expression levels of Apelin and Id1 in Wt and Heter ECs. The results were normalised to GAPDH. Cells were unstimulated (control (Con)) or stimulated with 1 ng·mL−1 tacrolimus (FK506) or 0.2 or 1 μmol·L−1 BUR1 in 0.1% serum for 1 h after serum-starvation for 16 h (n=3). e) Quantitative real-time PCR and immunoblotting results show the mRNA level of BMPR2 and protein levels of BMPRII, p-Smad1/5 and Id1 in BOECs from healthy donor (HD) and a patient with PAH normalised to GAPDH. Cells were unstimulated (Con) or stimulated with 0.2 μmol·L−1 BUR1 (quantitative real-time PCR) or 0.2 or 1 μmol·L−1 BUR (immunoblotting) in 0.1% serum for 4 h after starvation (n=3). f) Representative images of tube formation by BOECs from HD or PAH in various conditions. After serum-starvation for 16 h, a total of 20 000 cells was added to 48-well plates pre-coated with matrigel. The cells were stimulated with 30 ng·mL−1 vascular endothelial growth factor (VEGF) or 0.2 μmol·L−1 BUR1, in parallel with unstimulated cells as control (Con). The formation of tube-like structure was detected at 4 h post-seeding. Scale bars, 200 μm. g The number of junctions, h) number of branches and i) total length of branches in 10 fields of each assay were quantitatively analysed using ImageJ software (n=3). The BOECs of passages 4 and 6 were applied in these experiments. Black arrows indicate the 16 kDa dimer form of the Apelin protein. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 versus Con in the corresponding group; ##p<0.01, ###p<0.001, ####p<0.0001 versus HD Con.
BUR1 prevents and reverses MCT-induced PAH in rats
Next, we assessed whether BUR1 could prevent vascular remodelling in a PAH animal model [23]. As shown in figure 5a–d, 3 weeks of BUR1 treatment (4.5 mg·day−1, i.g.) initiated 1 week after MCT injection resulted in significantly reduced pulmonary arterial pressure and right ventricular systolic pressure (RVSP, 35±1.5 mmHg) compared with those of the saline-treated group (45±1.5 mmHg). Treatment with BUR1 also prevented right ventricular hypertrophy and thickening of the peripheral pulmonary artery in the MCT rat. Although preventative treatment with another antihypertensive agent, formononetin [24], reduced RVSP in the MCT–PAH model, the nonspecific reduction in mean systemic arterial pressure (MAP) excluded it as a potential treatment of PAH. Comparing the efficacy of daily administration of 0.45, 4.5 and 45 mg·kg−1 in rats (i.g.) revealed that the medium and high doses of BUR1 have similar effects on disease prevention and survival in the animal model without a change in systemic blood pressure (figure 5a–d).

BUR1 prevents and reverses monocrotaline (MCT)-induced pulmonary arterial hypertension. a) Assessment of right ventricular systolic pressure (RVSP), b) right ventricular hypertrophy (the ratio of right ventricular (RV) weight to left ventricular (LV) plus septal (S) weight (RV/(LV+S)) and c) mean arterial pressure (MAP) in rats on a prevention protocol given either vehicle (control (Con), n=6) or MCT (subcutaneously (s.c.)) for 1 week and subsequently treated with vehicle (MCT, n=6) or BUR1 (BUR1, intragastric (i.g.), n=6) from day 8 to 21. d) Pulmonary arterial wall thickness was quantified by the percentage of media wall thickness to diameter (diameter 50–100 μm, n=6 for each group, 10 fields for each slide). e) Assessment of RVSP, f) RV/(LV+S) and g) MAP in rats on a reversal protocol given either vehicle (Con) or MCT (55 mg·kg−1, s.c., n=18) and subsequently treated with saline (MCT, n=9) or BUR1 (BUR1, 4.5 mg·kg−1, n=9) from days 21–35 post-MCT; h) the muscularisation was analysed in pulmonary arteries with diameter 30–80 μm. i) Representative immunoblotting and densitometric analyses for the protein expression levels of phosphorylated Smad1/5 and Id1 in whole lung lysates from rats undergoing reversal treatment (n=3). j) Representative confocal images of immunofluorescence staining from five slides for BMPRII (green) and CD146 (red) from the lung sections of control (Con) and MCT-exposed rats treated with saline (MCT) or BUR1 (BUR1) following the reversal protocol (n=6). Arrows indicate the pulmonary arterial endothelial cells. Nuclei were counterstained with DAPI (blue). Scale bars, 20 μm. ns, not significant. *p<0.05, **p<0.01, ***p<0.001.
The administration of BUR1 for 2 weeks following 21 days of exposure to MCT confirmed the efficacy of BUR1 as a treatment to reverse MCT-induced damage (figure 5e–h). Significant decreases in RVSP and right ventricular hypertrophy were recorded. There were no significant differences in MAP or body weight between the MCT saline- and BUR1-treated MCT rats (data not shown). The muscularisation of the peripheral pulmonary artery was also partially reversed by BUR1 treatment (figure 5h). Immunoblotting and quantification of p-Smad1/5 and Id1 in whole lung cell lysates from control, MCT saline- and BUR1-treated MCT rats revealed that canonical BMP signalling was improved in the BUR1-treated group (figure 5i). BMPRII staining was detected in the CD146-positive endothelial cells; the downregulated BMPRII expression in MCT rats was rescued by BUR1 treatment (figure 5j). The improved BMPRII downstream signalling, anti-proliferation and reduced macrophage infiltration resulting from BUR1 treatment was further confirmed by immunostaining in pulmonary arteries of the BUR1-treated rats (supplemental figure S9). Assessing alkaline phosphate expression after chronic treatment, using BMP2 as a positive control, showed that BUR1 had no osteogenic effects on C2C12 cells (supplemental figure S10). Further pharmacological studies showed that BUR1 has low toxicity and a half-life of about 12.7 h in aqueous solution at 37 °C (supplemental figures S11 and S12).
BUR1 attenuates PAH in the SU/Hy rat model by enhancing BMP signalling
After the administration of one single dose of Sugen, rats were placed in conditions of chronic hypoxia for 3 weeks, followed by a 3-week period of normoxia. At the end of the normoxia period, SU/Hy-treated rats showed increased RVSP (97±12 mmHg) compared with the control group (34±5 mmHg). Over the following 3 weeks, 4.5 mg·kg−1 of BUR1 were administered daily. BUR1 reversed established PAH, including reductions in RVSP, mean pulmonary arterial hypertension and right ventricular hypertrophy (figure 6a–c). There were no significant differences in MAP between the SU/Hy and SU/Hy+BUR1 rats (figure 6d). Assessment of left ventricular function demonstrated that cardiac output and tricuspid annular plane systolic excursion (TAPSE) were improved in SU/Hy rats treated with BUR1 compared to vehicle (figure 6e, supplemental figure S13 and table S5). We also found that p-Smad1/5 and Id1 signalling were co-localised with CD31-positive endothelial cells in the pulmonary distal arteries. SU/Hy rats had reduced Id1 and p-Smad1/5 staining in obliterated pulmonary vasculature, which consisted of apoptosis-resistant endothelial precursor cells. Lastly, upregulated canonical BMP signalling was confirmed in the BUR1-treated group (figure 6f, g).

BUR1 attenuates Sugen 5416/hypoxia (SU/Hy)-induced pulmonary arterial hypertension. Rats were given vehicle (control (Con), n=6) or challenged with SU-5416 (subcutaneous (s.c.)) and maintained in hypoxia for 3 weeks then in normoxia for 3 weeks. After daily treatment with carboxymethyl cellulose vehicle (SU/Hy, n=6) or BUR1 (SU/Hy+BUR1, intragastric (i.g.), n=6) for another 3 weeks, the haemodynamic indexes of a) right ventricular systolic pressure (RVSP), b) mean pulmonary arterial hypertension (mPAP) and c) RV/(LV+S), plus d) mean arterial pressure (MAP) and e) cardiac output were assessed at 9 weeks in rats. f, g) Representative confocal images of immunofluorescence staining from five slides for Id1 (green) and CD31 (red) (f), and CD31 (red) and p-Smad1/5 (green) (g) from control, SU/Hy and SU/Hy+BUR1 lung sections (n=6). Nuclei were counterstained with DAPI (blue). White arrow indicates the localization of Id1 in the nucleus. Scale bars, 20 μm. ns, not significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
BUR1 modulates PTGS2 and arachidonate 5-lipoxygenase activating protein in pulmonary arterial endothelium
PTGS2 may be another important factor involved in BUR1-attenuated PAH. Another important regulator of the arachidonic acid metabolism pathway, arachidonate 5-lipoxygenase activating protein (ALOX5AP), showed an opposite expression trend when treated with BUR1 (figure 7a). Prolonged incubation with BUR1 for 24 h consistently altered PTGS2 and ALOX5AP protein levels from those recorded at earlier time points (figure 2f and figure 7b). The expression of both Ptgs2 and Alox5ap was increased in SU/Hy rat lung lysates (figure 7c, d) and in the thickened vasculature of SU/Hy rat lungs. Ptgs2 expression was maintained at a high level in the SU/Hy group, whereas the expression of Alox5ap was reduced to baseline levels by the treatment of BUR1 (figure 7e, f). The expression of PTGIS and TBXAS1, downstream enzymes in the PTGS2 cascade, was differentially regulated by BUR1 stimulation in the PAECs (supplemental figure S14a and S14b). Moreover, the expression of genes involved in LTB4 metabolism markedly diminished in the lungs of BUR1-treated animals compared to those of SU/Hy rats (supplemental figure S14c). To further understand the comprehensive modulation of eicosanoid metabolites, we measured PGE2 and LTB4 levels in the plasma of SU/Hy rats and those treated with BUR1. Although both were upregulated in SU/Hy rat plasma, LTB4 demonstrated a robust reduction on BUR1 treatment (figure 7g, h). Taken together, these results indicate that BUR1 modulated PTGS2 towards an anti-inflammatory effect in pulmonary vasculature.

BUR1 modulates PTGS2 and ALOX5AP in pulmonary arterial endothelial cells (PAECs) and rat lungs. a) Relative mRNA and protein expression of ALOX5AP in the PAECs after 4 h of treatment with different doses of BUR1; the densitometric analysis of protein expression is shown in the right panel (n=3). b) Representative immunoblotting shows the expression of PTGS2 and ALOX5AP in the PAECs after 24 h of treatment with different doses of BUR1; the densitometric analyses are shown in the right panels (n=3). c, d) Relative mRNA expression of Ptgs2 (c) and Alox5ap (d) in whole lung lysates from rats (n=3). e, f) Representative images of immunofluorescence staining from five slides for Ptgs2 (f, red) or Alox5ap (g, red) and CD31 (green) from control, SU/Hy and SU/Hy+BUR1 lung sections (n=6). Nuclei were counterstained with DAPI (blue). Scale bars, 20 μm. g) PGE2 and h) LTB4 levels in the plasma of SU/Hy rats and SU/Hy rats treated with BUR1 (n=3). The PAECs of passages 4 and 6 were applied in these experiments. ns: not significant. *p<0.05, ****p<0.0001.
Discussion
Previous reports have suggested increasing endothelial BMPRII signalling as a strategy to improve pulmonary vascular remodelling in PAH. Here, we report a new BMP2 regulator, BUR1, which regulates BMPRII signalling and the arachidonic acid pathway in PAECs. It effectively prevents and reverses PAH in rat models of disease. To provide an autologous and comparable model of heritable PAH, we also generated a disease-causing mutation in hESC-derived endothelial cells to assess the mechanisms of action of new small-molecule compounds.
BMP signalling is mediated by the formation of heteromeric receptor complexes of type II and type I receptors [25]. BMPRII signalling includes the canonical and non-canonical pathways, which are both affected by BMPRII deficiency in PAH. Canonical BMP signalling involves Id proteins, which function as downstream targets of Smad1/5/8 activation [26–28]. Id1 transcription is significantly increased by BMP2 in endothelial cells; therefore, we combined a primary screening for an upregulator of BMP2 with a secondary screening for an activator of the Id gene transcription in hESC-derived endothelial cells to selectively narrow the scope of the positive hits. We differentiated dual reporter lines into endothelial cells to test the compounds selectively.
BUR1 has a planar structure with a pyrazolopyrimidine core. The closest structure reported is that of the {1H-pyrazolo[3,4-d]pyrimidin-4-yl}piperidine compound, which functions as a serine–threonine and tyrosine kinase modulator for the treatment of immunological, inflammatory and proliferative diseases. BUR1 has the lowest EC50 of the examined analogues for upregulation of BMP2 and Id1 transcription. We considered whether BUR1 has the same action as the pyrimidinone core of sildenafil, which competitively blocks the hydrolysis of cyclic guanosine monophosphate by phosphodiesterase type 5 (PDE5). However, although these compounds share structural similarities, BUR1 demonstrates a much lower inhibitory effect on PDE5A1, showing a median inhibitory concentration (IC50) >20 μmol·L−1. Thus, it is possible that BUR1 acts via the tyrosine kinase to modulate BMP signalling. We also investigated the off-target effects of BUR1 on the osteogenesis of C2C12 cells and showed that BUR1 did not induce alkaline phosphatase activity to the same degree as BMP2.
BUR1 demonstrated its potential as a regulator of BMPRII signalling with an EC50 of about 0.098 μmol·L−1 in the hESCs-Id1-Venus-Luc reporter system. Moreover, BUR1 enhanced BMP2 and PTGS2 expression in endothelial cells. We also found that a low dose of BUR1 effectively increased Smad1/5 activation and Id1 transcription in PAECs. The sequential drug discovery went from standard screening with a micromolar level down to the selective and effective dosage of BUR1 (0.2 μmol·L−1). At early time points, BUR1 affects BMPRII signalling. At later time points, the indirect effects on PTGS2 and other factors become apparent, which could be beneficial for long-term treatment. We found that BMP2 was consistently upregulated from early time points to 24 h, when the conveyed signal enhanced the expression of BMPRII. We further investigated the mechanism by which BUR1 activates BMPRII signalling at early time points by using LDN-193189 to block type I and II receptors in the presence of 0.1% serum. We found that canonical BMP signalling was diminished in a dose-dependent manner. Our findings indicate a dose-dependent blockade of LDN-193189 on BUR1 activation in the presence of BMP9. We used a specifically designed siRNA to knockdown BMPRII and ActRII receptors, and the reduced levels of Id1 implicated both of these receptors as components in BUR1-stimulated canonical BMP signalling. A promoter deletion assay further clarified that BUR1 regulates BMP2 transcription by enhancing Smads binding on its promoter.
We employed the CRISPR/Cas9 genetic targeting tool to generate the R899X point mutation in hESCs. This model system has a number of benefits. First, this system provides an unlimited, expandable resource of human BMPR2+/R899X endothelial cells. Although puromycin completely recovered the mutant transcript in cells obtained from the BMPR2+/R899X mouse, it exhibited only a partial effect in human cells [15]. Second, this system generates an autologous counterpart mutant to minimalise the variation between biological individuals. Finally, it is possible to study the effects of the genetic factors on the development of human subjects. A recent study demonstrated that induced pluripotent stem cells from induced/heritable PAH differentiated into endothelial cells have the same gene expression profile as PAECs from the same patient [29]. The remaining BMPRII maintained the basal activation of canonical BMP signalling via Smads by BUR1. We confirmed an elevation of BMPRII signalling using the same treatment in endothelial cells from PAH patients with BMPRII deficiencies.
We found significantly increased PTGS2 expression and simultaneously reduced ALOX5AP expression in BUR1-treated endothelial cells and SU/Hy rat pulmonary arteries. We also found increased PTGS2 expression could be induced by BUR1 treatment via BMP2 in PAECs. BMP2 has been reported to activate activating transcription factor-2 and runt-related transcription factor-2 to bind to the proximal promoter of PTGS2 [30]. Meanwhile, BUR1 treatment reduced LTB4 production, owing to the reduced expression of ALOX5AP resulting in less oxidation of arachidonic acid. We also detected a mild increase in PGE2 production as a vasodilator in the plasma from treated SU/Hy rats. PGE2 is a critical mediator of inflammation that induces both pro- and anti-inflammatory effects and signals via four different signalling-E prostanoid (EP) receptors, EP1 to EP4 [31, 32]. PGE2 exhibits various effects in the lungs, such as pulmonary vascular remodelling and inflammatory regulation [33]. In this study, the balance of arachidonic acid metabolism in the PAECs was tipped by BUR1 to favour PTGS2 rather than ALOX5AP activity, leading to an elevated PGE2/LTB4 ratio in the pulmonary endothelium from BUR10-treated PAH rats. These results indicate that BUR1 modulates PTGS2 to favour an anti-inflammatory response in experimental PAH (figure 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed stem cell-based drug screening and validation system. BUR1, a piperidine, was identified by Id1-Venus-Luc dual reporter human embryonic stem cell (hESC)-derived endothelial cells (ECs) as a BMPRII signalling regulator from among 31 small-molecule compounds. In vitro, BUR1 enhanced the transcription of BMP2 and, later, PTGS2 in pulmonary arterial ECs. Its effect was validated in the autologous heritable pulmonary arterial hypertension (HPAH) mutation BMPR2+/R899X hESC-ECs and in idiopathic PAH blood outgrowth ECs. Both the Smad-dependent pathway (Id) and non-Smad-dependent pathway (e.g. Apelin) were activated, promoting angiogenesis in the BMPRII-deficient ECs. The therapeutic effects of BUR1 for PAH were further validated in the monocrotaline (MCT)–PAH rat model and Sugen 5416/hypoxia (SU/Hy)–PAH rat model in vivo. This work indicates a new system for stem cell-based drug screening.
Our in vivo studies demonstrated the potential for BUR1 to act as a therapeutic agent for PAH. We tested three different doses of BUR1 in a prevention study using the MCT rat model and selected the dose of 4.5 mg·kg−1·day−1, which proved to be efficient in halting the progression of PAH. The reversal of established PAH in MCT rats was achieved via treatment with BUR1, which reduced RVSP and medial hypertrophy following the administration of MCT. We also demonstrated the restoration of BMPRII signalling in the pulmonary arterial wall of BUR1-treated rats. A more promising result of this study was obtained when examining chronic hypoxia together with the Sugen induced rat model of PAH, and following haemodynamic tests including echocardiography; in this experiment, BUR1 not only attenuated phosphorylated Smad1/5 and Id1 protein levels, but also regulated the balance of PTGS2 and ALOX5AP expression in the neointimal lesion, which indicates that targeting BMPRII signalling to potentiate anti-inflammatory effects is a valid therapeutic strategy for PAH.
In conclusion, this study provides the basis for the development of a therapeutic strategy for PAH that targets BMPRII signalling. Using a stem cell reporter line, we identified a selective BMP2 upregulator in endothelial cells. The enhanced BMPRII signalling by BUR1 was observed not only in PAECs but also in autologous mutant hESC endothelial cells and BOECs obtained from PAH patients. BUR1 also pushes arachidonic acid metabolism towards PTGS2-driven anti-inflammatory effects. Using the MCT and SU/Hy PAH animal models has allowed us to provide a novel structural type for the treatment of PAH and other related diseases (figure 8).
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02229-2017_supp
Acknowledgments
We would like to thank Mariaestela Ortiz (Wellcome Trust-MRC Cambridge Stem Cell Institute, Cambridge, UK) for designing the dual reporter line and Zhongqiu Chen (Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China) for making the promoter partial deletion constructs. We also thank Xiaojian Wang (State Key Laboratory of Cardiovascular Disease, FuWai Hospital, Beijing, China) for help with the animal experiments.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: J. Yang designed the research studies and finalised the manuscript; Y. Xing conducted the animal experiments and PTGS2-related study, and revised the manuscript; Q. Wei, S. Zhao, F. Zhou and R. Al-Lamki conducted the experiments and acquired the data; G. Shang performed the chemical screening; X. Zhao performed the immunoblotting experiment; D. Ortmann and M. Du performed the stem cell experiments; R. Pedersen provided stem cell-related reagents; S. Si provided chemicals; N.W. Morell revised the manuscript and provided cell culture reagents.
Support statement: This work was supported by the National Key Research and Development Program of China – Stem Cell and Translational Research (number 2016YFA0102300), CAMS Innovation Fund for Medical Sciences (CIFMS 2016-I2M-4-003), National Natural Science Foundation of China (NSFC) (81400278 and 81670054), China National Thousand (Young) Talents Program and The Fundamental Research Funds for the Central Universities to J. Yang; NSFC (81621064) to S. Si; and the International Postdoctoral Exchange Fellowship Program to Y. Xing. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: None declared.
- Received October 31, 2017.
- Accepted February 10, 2018.
- Copyright ©ERS 2018
References










