





Abstract
We evaluated the efficacy and safety of ciprofloxacin dry powder for inhalation (DPI) in patients with non-cystic fibrosis bronchiectasis, two or more exacerbations in the previous year and predefined sputum bacteria.
Patients were randomised 2:1 to twice-daily ciprofloxacin DPI 32.5 mg or placebo in 14- or 28-day on/off treatment cycles for 48 weeks. Primary end-points were time to first exacerbation and frequency of exacerbations. Enrolling countries and α level split (0.049 and 0.001 for 14- and 28-day cycles, respectively) differed from RESPIRE 1.
Patients were randomised to ciprofloxacin DPI (14 days on/off (n=176) or 28 days on/off (n=171)) or placebo (14 days on/off (n=88) or 28 days on/off (n=86)). The exacerbation rate was low across treatment arms (mean±sd 0.6±0.9). Active treatment showed trends to prolonged time to first exacerbation (ciprofloxacin DPI 14 days on/off: hazard ratio 0.87, 95.1% CI 0.62–1.21; p=0.3965; ciprofloxacin DPI 28 days on/off: hazard ratio 0.71, 99.9% CI 0.39–1.27; p=0.0511) and reduced frequency of exacerbations (ciprofloxacin DPI 14 days on/off: incidence rate ratio 0.83, 95.1% CI 0.59–1.17; p=0.2862; ciprofloxacin DPI 28 days on/off: incidence rate ratio 0.55, 99.9% CI 0.30–1.02; p=0.0014), although neither achieved statistical significance. Ciprofloxacin DPI was well tolerated.
Trends towards clinical benefit were seen with ciprofloxacin DPI, but primary end-points were not met.
Abstract
RESPIRE 2 supports ciprofloxacin DPI clinical benefits in bronchiectasis, but did not reach statistical significance http://ow.ly/es9E30gNhos
Introduction
Non-cystic fibrosis bronchiectasis (NCFB) is a heterogeneous disease with a range of underlying aetiologies across geographical regions and a variable clinical course [1]. Patients with NCFB are subject to a vicious cycle of inflammation, poor mucus clearance and recurrent infection [2, 3]. This cycle manifests as a chronic cough, sputum production, airway damage and exacerbations, with the severity and impact of each exacerbation varying from patient to patient, and is responsible for progressive lung damage [2, 4].
Exacerbations of bronchiectasis are characterised by a debilitating increase in symptoms and are associated with poorer health outcomes, including reduced quality of life, depression, anxiety and increased mortality [5–7]. Reducing the frequency of exacerbations is therefore an important treatment goal; however, there are currently no antibiotic therapies licensed for this.
The RESPIRE programme consisted of two trials of the same overall design, as required by regulatory agencies. The objective of both studies was to assess the efficacy and safety of 14- and 28-day on/off regimens of ciprofloxacin dry powder for inhalation (DPI) in prolonging time to first exacerbation and reducing frequency of exacerbations in patients with NCFB. The RESPIRE 1 trial demonstrated that treatment with a 14-day on/off cycle of ciprofloxacin DPI significantly reduced frequency of exacerbations and prolonged time to first exacerbation in patients with NCFB. The 28-day ciprofloxacin DPI arm of RESPIRE 1 had trends towards clinical benefit, but failed to reach statistical significance [8]. Here, we report the results of the phase III RESPIRE 2 trial (ClinicalTrials.gov identifier NCT02106832). The main differences between RESPIRE 1 and 2 were a change in the recruiting countries (RESPIRE 2 drew more patients from Asia and Eastern Europe than RESPIRE 1) and specific alterations to the statistical plan, as described in the following sections.
Methods
Study design and treatment
The RESPIRE trials had the same overall methodology, which has been reported previously [9]. Two interventions were studied over 48 weeks: ciprofloxacin DPI 32.5 mg twice daily 14 days on/off (12 cycles) and ciprofloxacin DPI 32.5 mg twice daily 28 days on/off (6 cycles). There was an 8-week off-treatment follow-up period.
Study population
Patients with a primary diagnosis of NCFB (idiopathic or post-infectious aetiology) and stable disease were enrolled. Key inclusion criteria were: at least two exacerbations in the previous 12 months and a positive sputum culture at screening for at least one of seven pre-specified pathogens (Pseudomonas aeruginosa, Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus, Streptococcus pneumoniae, Stenotrophomonas maltophilia or Burkholderia cepacia). Patients with active allergic bronchopulmonary aspergillosis, active or actively treated nontuberculous mycobacterial lung infection or tuberculosis, a primary diagnosis of chronic obstructive pulmonary disease (COPD), or documented chronic asthma were excluded. Randomisation was stratified by geographical region, pre-therapy positive culture for P. aeruginosa and chronic macrolide use.
End-points
Two different hierarchical statistical analysis approaches were developed with different regulatory authorities. The primary end-point for the US Food and Drug Administration (FDA) was time to first exacerbation within 48 weeks after start of treatment (ciprofloxacin DPI versus pooled placebo). For the European Medicines Agency (EMA)/other agencies, such as the China State Drug Administration, the primary end-point was frequency of exacerbations during the 48-week study (ciprofloxacin DPI versus matching placebo). The pooled placebo group refers to all patients from the 14- and 28-day on/off placebo arms; matching placebo refers to ciprofloxacin DPI 14 days on/off compared with placebo 14 days on/off and ciprofloxacin DPI 28 days on/off compared with placebo 28 days on/off [4]. For the primary end-points, exacerbations had to meet three criteria: 1) worsening of at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variations for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue and 3) systemic antibiotic treatment (“primary end-point definition”). Pre-defined subgroups and safety end-points are described in supplementary sections S1 and S2). Secondary end-points have been detailed previously [8, 9]. A secondary end-point of particular interest was exacerbation rate according to a less stringent definition, i.e. a respiratory event with worsening of at least one of the aforementioned signs or symptoms and systemic antibiotic use.
Analyses
The statistical analysis plan for the RESPIRE programme has been described previously [9]. RESPIRE 2 followed the same analysis plan as for RESPIRE 1 with two exceptions. First, the primary analysis models for the frequency of exacerbations end-point were harmonised between the FDA and EMA/others analyses with respect to the handling of treatment discontinuations. In the RESPIRE 1 FDA analysis, the frequency of exacerbations was extrapolated for patients not completing the 48-week period, whereas in RESPIRE 2 the patients' time in study was included as an offset variable in the FDA analysis as for the EMA/others analysis. Second, the significance levels (α level split) were amended before study end, based on experience gained from RESPIRE 1. In RESPIRE 1, a Bonferroni correction was used, resulting in a significance level of 0.025 for each treatment arm (14 and 28 days on/off). Following the statistical failure of the ciprofloxacin DPI 28-day on/off arm in RESPIRE 1, the α correction for RESPIRE 2 was prospectively amended to a weighted Bonferroni correction of 0.049 for the 14-day on/off arm and 0.001 for the 28-day on/off arm. The sponsoring company discussed this change before study end with the regulatory agencies who approved this change. Further details are given in supplementary section S3.
Results
Patients
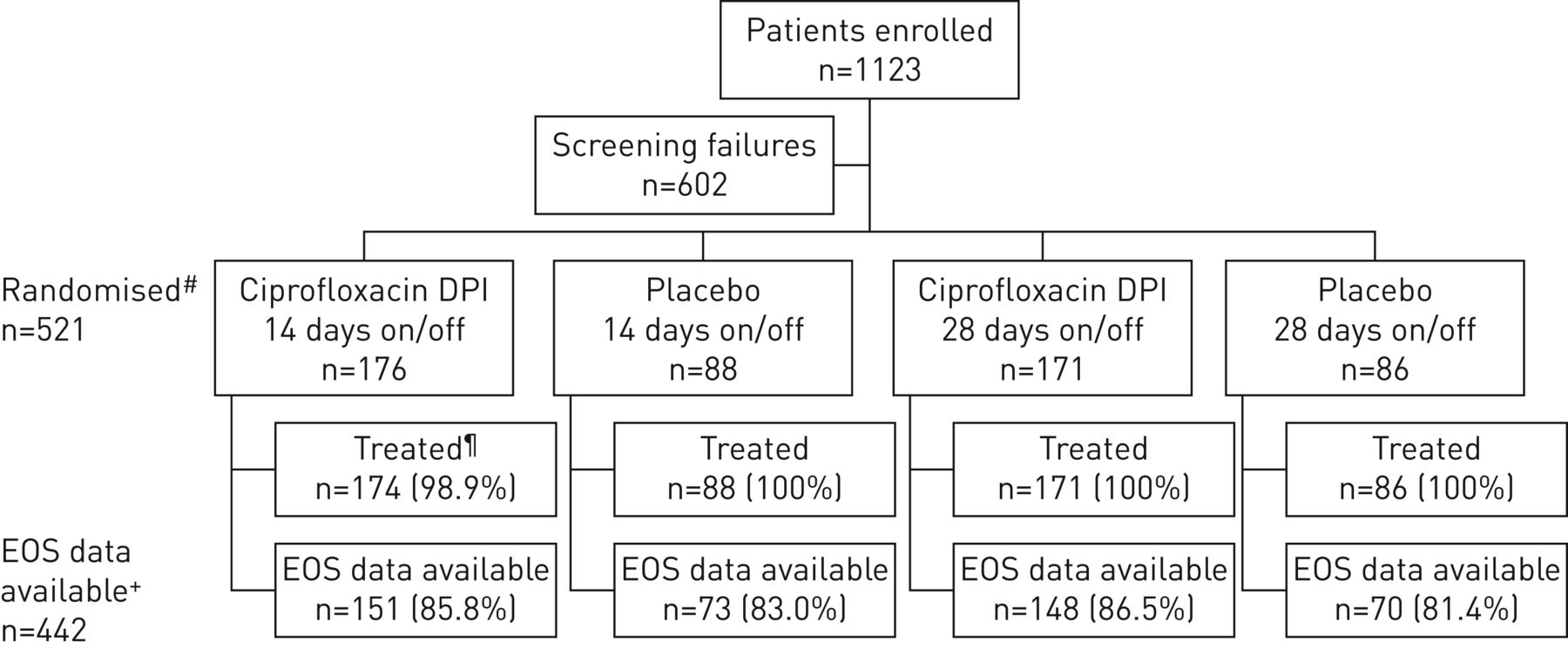
A total of 521 patients from 24 countries were randomised to treatment (figure 1); the first patient visit was in April 2014 and the last visit was in October 2016. The highest enrolling countries were Russia (11.5%), Bulgaria (10.4%) and Latvia (8.6%) (supplementary table S1).

Patient disposition. DPI: dry powder for inhalation; EOS: end of study. #: randomisation stratified by geographical region, pre-therapy positive culture for Pseudomonas aeruginosa and chronic macrolide use; ¶: patients randomised who received at least one treatment dose; +: patients with EOS data available include those who completed treatment and subsequent follow-up, or prematurely discontinued treatment but were followed further until the planned EOS (52/54 weeks), or prematurely discontinued treatment but completed the planned 8 weeks of follow-up.
Patient demographics and baseline characteristics were balanced across groups (table 1). The majority of patients (58.0%) were female, the mean age was ∼60 years and patients had a high likelihood of P. aeruginosa infection (60.7% of patients; see supplementary table S2 for additional pre-specified baseline pathogens). Lung function was generally poor, with >40% of patients having a forced expiratory volume in 1 s (FEV1) % pred <50%. Approximately 30% of patients had a medical history of COPD and use of concomitant respiratory medications considered part of NCFB therapy was common (supplementary table S3).
Patient demographics and baseline characteristics
442 patients (84.8%) completed the study and 79.5% of patients (414 out of 521) completed treatment. Patient compliance was high (mean >95% in all arms) (table 2).
Treatment compliance and duration#
Primary end-points
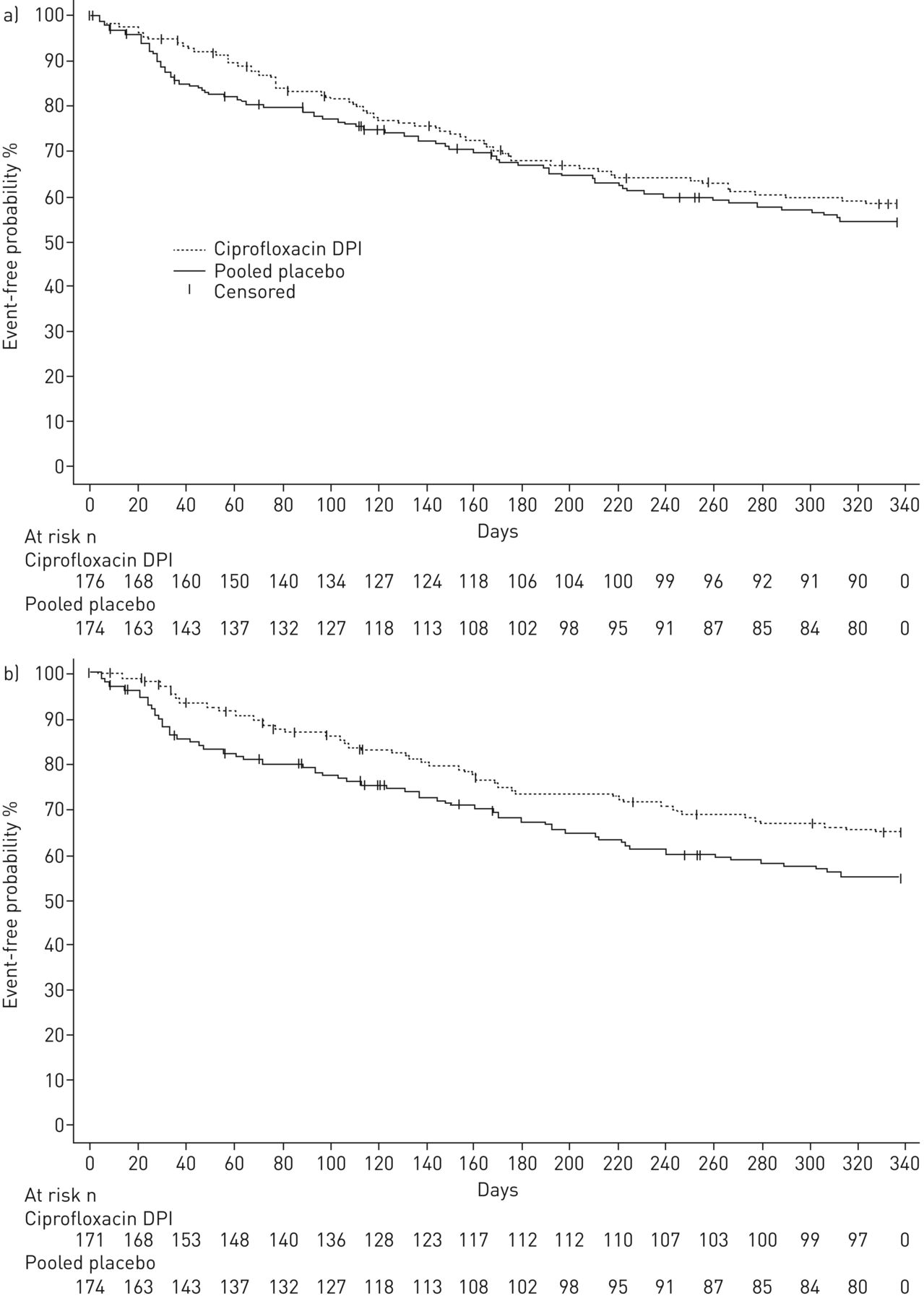
Both the 14- and 28-day ciprofloxacin DPI on/off arms increased the time to first exacerbation versus pooled placebo (figure 2). However, neither ciprofloxacin DPI treatment arm demonstrated a statistically significant difference compared with pooled placebo (ciprofloxacin DPI 14 days on/off: hazard ratio (HR) 0.87, 95.1% CI 0.62–1.21; p=0.3965 and ciprofloxacin DPI 28 days on/off: HR 0.71, 99.9% CI 0.39–1.27; p=0.0511) (table 3). The median time to first exacerbation was not estimable for any treatment arm due to the low exacerbation rates. Overall, 61.4% and 67.3% of patients treated with ciprofloxacin DPI 14 and 28 days on/off, respectively, did not experience an exacerbation matching the stringent definition, compared with 58.0% of patients in the pooled placebo arm.

Time to first exacerbation for patients receiving ciprofloxacin dry powder for inhalation (DPI) a) 14 days on/off or placebo and b) 28 days on/off or placebo. For the primary end-point exacerbations were required to meet three criteria: 1) worsening in at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variation for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue, and 3) systemic antibiotic treatment.
Primary end-points: time to first exacerbation and frequency of exacerbations# in patients treated with ciprofloxacin dry powder for inhalation (DPI) versus placebo over 48 weeks
Ciprofloxacin DPI 14 days on/off reduced the exacerbation incidence rate by 17% over 48 weeks (incidence rate ratio (IRR) 0.83, 95.1% CI 0.59–1.17; p=0.2862), while ciprofloxacin DPI 28 days on/off reduced the exacerbation incidence rate by 45% (IRR 0.55, 99.9% CI 0.30–1.02; p=0.0014) (table 3). Neither arm reached the predefined statistical significance level (p=0.049 for the 14-day regimen and 0.001 for the 28-day regimen). Overall, the frequency of exacerbations in all arms was very low (figure 3). The overall mean±sd number of exacerbations was 0.6±0.9. Equivalent data for the treatment arms were 0.6±0.8 for ciprofloxacin DPI 14 days on/off compared with 0.7±1.0 in the matching placebo arm and 0.4±0.6 for ciprofloxacin DPI 28 days on/off versus 0.7±1.1 in the matching placebo arm.

Descriptive frequency of exacerbations over 48 weeks for patients receiving ciprofloxacin dry powder for inhalation (DPI) a) 14 days on/off or matching placebo and b) 28 days on/off or matching placebo. Numbers within or above bars indicate number of patients. For the primary end-point exacerbations were required to meet three criteria: 1) worsening in at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variation for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue, and 3) systemic antibiotic treatment.
Over 48 weeks, 61.4% of patients treated with ciprofloxacin DPI 14 days on/off compared with 67.3% of patients treated with ciprofloxacin DPI 28 days on/off and 58.0% of patients in the pooled placebo arm did not experience a protocol-defined exacerbation for the primary end-point.
Subgroup analyses
Exploratory analyses of the primary end-point in pre-specified subgroups did not reveal any differences in any of the subgroups for time to first exacerbation or frequency of exacerbations (supplementary figure S1).
Secondary end-points
Due to the hierarchical nature of the statistical analysis plan, formal significance testing could not be performed for secondary end-points because the primary end-points were not met. Exploratory analyses of secondary end-points suggested that ciprofloxacin DPI treatment was associated with benefits, including a reduced frequency of exacerbations (defined according to the primary end-point) versus pooled placebo for the ciprofloxacin DPI 28-day on/off arm (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Results for ciprofloxacin dry powder for inhalation (DPI) in US Food and Drug Administration (FDA) and European Medicines Agency (EMA)/other agencies hierarchical analyses for ciprofloxacin DPI a) 14 days on/off and b) 28 days on/off versus pooled placebo (PPL) or matching placebo (MPL). HR: hazard ratio; IRR: incidence rate ratio; EOT: end of treatment; SGRQ: St George's Respiratory Questionnaire; QOL-B RSS: Quality of Life-Bronchiectasis respiratory symptoms domain score; FEV1: forced expiratory volume in 1 s. #: the primary (stringent) definition of exacerbation was a respiratory event that met three separate criteria: 1) worsening in at least three signs or symptoms (dyspnoea, wheezing, cough, 24-h sputum volume or sputum purulence) beyond normal day-to-day variation for at least 2 consecutive days, 2) fever (body temperature >38.0°C) or malaise/fatigue, and 3) systemic antibiotic treatment; ¶: the less stringent definition of exacerbation was a respiratory event with worsening of at least one of the aforementioned signs or symptoms and systemic antibiotic use; +: Pseudomonas aeruginosa, Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus, Streptococcus pneumoniae, Stenotrophomonas maltophilia or Burkholderia cepacia (eradication of pathogens was defined as a negative culture result at EOT for all pre-specified pathogens in a subject with a positive baseline culture for at least one pre-specified pathogen; occurrence of a new pathogen was defined as a positive culture for at least one pre-specified pathogen at EOT in a subject who did not have a positive culture for that pathogen at baseline). Dashed lines indicate statistical significance was not reached. The α level for end-points was 0.049 for the 14-day arm and 0.001 for the 28-day arm. The confidence interval is 95.1% for comparisons of ciprofloxacin DPI 14 days on/off versus placebo and 99.9% for comparisons of ciprofloxacin DPI 28 days on/off versus placebo.
The mean±sd number of exacerbations reported using the less stringent definition was 0.7±1.0 for ciprofloxacin DPI 14 days on/off compared with 0.8±1.1 in the matching placebo arm and 0.5±0.8 for ciprofloxacin DPI 28 days on/off compared with 0.9±1.1 in the matching placebo arm.
Safety
Ciprofloxacin DPI was well tolerated, with lowest adverse event rates observed in the ciprofloxacin DPI 28-day on/off arm. A total of 349 out of 519 (67.2%) patients experienced an adverse event at any time after enrolment: 131 (75.3%) patients in the ciprofloxacin DPI 14-day on/off arm, 101 (59.1%) in the ciprofloxacin DPI 28-day on/off arm and 117 (67.2%) in the pooled placebo group.
Overall, 64.9% of patients experienced a treatment-emergent adverse event. Treatment-emergent musculoskeletal adverse events were infrequently reported. Tendon disorders were reported by two patients in each ciprofloxacin DPI treatment group (one tendon disorder and one trigger finger in the ciprofloxacin DPI 14-day on/off arm; discomfort and one ligament sprain in the ciprofloxacin DPI 28-day on/off arm) and zero patients in the placebo groups. Premature discontinuations due to treatment-emergent adverse events were uncommon and occurred with similar frequencies across treatment arms (table 4).
Treatment-emergent adverse events (TE-AEs)# (MedDRA classifications)
Serious treatment-emergent adverse events occurred in 22.0% of patients overall; rates were slightly lower in the ciprofloxacin DPI 28-day on/off arm compared with other arms. Respiratory disorders and infections/infestations were the most common system organ classes reported for serious treatment-emergent adverse events (supplementary table S4). One serious treatment-emergent adverse event (atrial flutter) was considered drug related. This treatment-emergent adverse event occurred in a subject in the ciprofloxacin DPI 28-day on/off arm who had a history of ischaemic heart disease. This patient also experienced severe fatal congestive cardiomyopathy, which was reported in parallel but was not considered to be drug related.
Nine treatment-emergent deaths were reported during the study (three (1.7%) in the ciprofloxacin DPI 14-day on/off arm, four (2.3%) in the ciprofloxacin DPI 28-day on/off arm and two (1.1%) in the pooled placebo group), but no fatalities were considered to be drug related and deaths were consistent with morbidities common to the patient population being studied (supplementary table S4).
Changes in ciprofloxacin minimal inhibitory concentrations
At baseline, 98 out of 521 (18.8%) patients had a pathogen with an elevated minimal inhibitory concentration (MIC) for ciprofloxacin (defined as resistant based on systemic ciprofloxacin breakpoints as specified in supplementary section S2) isolated from their sputum. The number of patients with the development of at least one isolate from sputum with an elevated MIC from pre-treatment to any time point during the study was 37 out of 176 (21.0%) for ciprofloxacin DPI 14 days on/off, 28 out of 170 (16.5%) for ciprofloxacin DPI 28 days on/off and 17 out of 173 (9.8%) for pooled placebo. At the end-of-study (EOS) visit that occurred 8 weeks after the end of treatment, pathogens with elevated MICs were isolated from 26 patients (5%) (ciprofloxacin DPI 14 days on/off 12 out of 176 (6.8%), ciprofloxacin DPI 28 days on/off 10 out of 171 (5.8%) and pooled placebo four out of 174 (2.3%)) who had susceptible bacteria at baseline. During the study, elevated MICs were more common in patients receiving active therapy. The percentage of patients with at least one isolate from sputum with an elevated MIC at any time point including baseline and EOS was 52.3% for ciprofloxacin DPI 14 days on/off, 39.8% for ciprofloxacin DPI 28 days on/off and 29.3% for pooled placebo.
Discussion
The RESPIRE 2 trial constitutes the second trial in the RESPIRE programme. RESPIRE 1 demonstrated a significant treatment effect for the ciprofloxacin DPI 14-day on/off treatment regimen and a trend towards improved outcomes in the ciprofloxacin DPI 28-day on/off arm [8]. In RESPIRE 2, end-points favoured ciprofloxacin DPI over placebo, but the positive efficacy signals did not translate into statistical significance for either active treatment arm. The observed positive trends in RESPIRE 2 were generally more pronounced in the ciprofloxacin DPI 28-day on/off arm than in the 14-day on/off arm. However, due to the pre-specified significance level of 0.001 for the ciprofloxacin DPI 28-day on/off arm, differences from placebo did not achieve statistical significance.
Exacerbation rates in RESPIRE 2 were much lower than expected and lower than those observed in RESPIRE 1. In RESPIRE 2, 62.2% of patients did not experience an exacerbation according to the primary definition (defined by three signs or symptoms), which was notably higher than the 52% of patients in RESPIRE 1 with no exacerbations [8]. As the study entry criteria were chosen to enrich the population of “frequent exacerbators”, requiring two or more documented exacerbations in the previous year, the low exacerbation rates that occurred during the trial were unexpected. For both trials, a stringent definition of exacerbation was used for primary end-point evaluations [9]. This definition was not, however, applied for the entry criteria. As each patient experiences their disease differently [10] and as patients are often educated to take antibiotic treatment when symptoms increase, it is likely that the prior exacerbations did not meet the stringent definition used during the trial. A proposed standardised definition of an exacerbation has recently been published [11]; this may be of benefit in future clinical trials to reduce heterogeneity.
Understanding the low exacerbation rates observed in both active and placebo arms of RESPIRE 2, and the differences in exacerbation rates between RESPIRE 1 and 2, requires examination of a number of factors, including patient characteristics and geography. Baseline demographics varied somewhat between RESPIRE 1 and RESPIRE 2, despite their identical inclusion and exclusion criteria. As patient phenotype is known to affect prognosis [10], it is possible that the differing patient profiles led to different clinical outcomes. Patients in RESPIRE 2 were generally younger (mean age 60.1 versus 64.7 years) and more likely to have had only two exacerbations in the last year (78% versus 55%) than patients in RESPIRE 1 [8]; two episodes was the minimum frequency of exacerbations required to enter the study. RESPIRE 2 patients had poorer lung function as indicated by the proportion of patients with FEV1 % pred <50% (41.8% versus 30.0%) and more respiratory symptoms as assessed by the St George's Respiratory Questionnaire symptoms component score (mean 61.0 versus 56.8) [8].
Although a primary diagnosis of COPD was an exclusion criterion, 28% of patients in RESPIRE 2 had a history of COPD versus 16% in RESPIRE 1, suggesting that more patients in RESPIRE 2 had an overlapping diagnosis of bronchiectasis/COPD. This is supported by the greater theophylline use in RESPIRE 2 (8.1%) versus RESPIRE 1 (2.2%). The precise nature of the relationship between COPD and bronchiectasis remains to be established. Optimal treatment of these patients is poorly understood and may differ from therapies recommended for either condition alone [12]. Excluding patients with an overlapping bronchiectasis/COPD diagnosis may result in more homogeneous NCFB patient populations in clinical trials. Patients in RESPIRE 2 had indicators of more severe airflow obstruction, but bronchiectasis severity could not be fully evaluated because computed tomography scans were not assessed at baseline. Not all parameters included in bronchiectasis severity indices were collected at baseline, as details for these had not been published when the RESPIRE studies were being designed [7, 13].
RESPIRE 1 and 2 also differed in the geographical regions that were represented, with RESPIRE 2 enrolling more patients from Asia and Eastern Europe. The variability in clinical practice could have potentially impacted the determination of exacerbations and treatment prior to enrolment. Specialist care for individuals with bronchiectasis is well developed, and research interest is relatively intense in Western Europe, the USA and Australia, which has led to the development of treatment guidelines and national standards [14–16]. European guidelines have only recently been published [17] and similar initiatives have been less common in other areas of the world [1]. This may have led to different disease management practices prior to enrolment. For example, patients in RESPIRE 2 were less likely to be treated with standard respiratory medications (67.9% versus 80.5%) or long-term macrolide treatment (7.7% versus 15.9%) than those in RESPIRE 1 [8]. This might reflect individual country practices or could be explained by different patient phenotypes. Reducing the heterogeneity of patient populations, in terms of phenotypic differences and standards of care prior to enrolment, is an important consideration for future trials.
Any comparison of the outcomes of RESPIRE 1 and 2 should take into consideration the change in extrapolation method and significance levels. Based on experience gained in RESPIRE 1, significance levels were amended by the sponsor with agreement from regulatory authorities to maximise the likelihood of achieving a significant result in the ciprofloxacin DPI 14-day on/off arm. If the α adjustment applied in RESPIRE 1 (p=0.025) had been used in RESPIRE 2, the ciprofloxacin DPI 28-day on/off arm would have met the primary end-point for the EMA/others analysis of frequency of exacerbations (IRR 0.55, 99.9% CI 0.30–1.02; p=0.0014). The FDA primary end-point, however, would not have been statistically significant nor would results in the ciprofloxacin DPI 14-day on/off arm have been affected.
As observed in RESPIRE 1, ciprofloxacin DPI was well tolerated. The number of study-drug-related treatment-emergent adverse events was markedly lower in RESPIRE 2 than RESPIRE 1 (13.7% versus 25.1%) [8]. Differences were largely due to lower numbers of respiratory treatment-emergent adverse events; contributory factors may include a lower rate of exacerbations in RESPIRE 2 or a higher threshold for diagnosis of these adverse events in centres involved in RESPIRE 2 versus RESPIRE 1. Trial discontinuation rates were low (5% due to treatment-emergent adverse events) and comparable across treatment arms. Together with data from RESPIRE 1, these findings support a favourable safety profile for ciprofloxacin DPI.
The limitations of the RESPIRE programme study design have been discussed previously [8]. RESPIRE 2 additionally highlighted the impact of low exacerbation rates on efficacy outcomes. Future NCFB trials should consider using a more stringent definition for prior exacerbations as an inclusion criterion and collecting data needed to calculate bronchiectasis severity based on published severity assessment tools [7, 13].
In conclusion, despite efficacy signals showing trends towards clinical benefits and excellent tolerability in the RESPIRE 2 study, neither the 14- or 28-day on/off ciprofloxacin DPI arms met the primary end-points of increasing time to first exacerbation or reducing frequency of exacerbations. The RESPIRE clinical trial programme thus encapsulates both the promise and difficulties of documenting a statistically significant impact on exacerbations in NCFB patients, and substantially contributes to the body of knowledge on the optimal design of clinical trials.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02053-2017_Supplement
Disclosures
Supplementary Material
T. Aksamit ERJ-02053-2017_Aksamit
T-J. Bandel ERJ-02053-2017_Bandel
A. De Soyza ERJ-02053-2017_De_Soyza
J.S. Elborn ERJ-02053-2017_Elborn
E. Operschall ERJ-02053-2017_Operschall
E. Polverino ERJ-02053-2017_Polverino
K. Roth ERJ-02053-2017_Roth
R. Wilson ERJ-02053-2017_Wilson
K.L. Winthrop ERJ-02053-2017_Winthrop
Acknowledgements
We thank the patients, investigators (supplementary section S4) and study centres that contributed to RESPIRE 2. We also thank Ulrike Krahn (Bayer AG, Wuppertal, Germany) and Elaine Montegriffo (Bayer PLC, Reading, UK) for assisting with statistical analysis; Jeff Alder (Bayer US LLC, Whippany, NJ, USA) for microbiology data assessment and critically reviewing the data; Maxine Lau (Bayer AG, Berlin, Germany) for pharmacovigilance input; and Fusion MD (Montreal, Canada) and highfield:communication (Oxford, UK) for providing medical writing services with funding from Bayer AG. A. De Soyza, R. Wilson and J.S. Elborn acknowledge the support of EMBARC and the Medical Research Council-funded BRONCH-UK collaborative for peer support and advice in bronchiectasis.
Footnotes
This article has supplementary material available from erj.ersjournals.com
This study is registered at ClinicalTrials.gov with identifier NCT02106832.
Support statement: This study was supported by Bayer AG. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received October 6, 2017.
- Accepted November 20, 2017.
- Copyright ©ERS 2018
References










