





Abstract
Oxidative stress contributes to chronic obstructive pulmonary disease (COPD) exacerbations and antioxidants can decrease exacerbation rates, although we lack data about the effect of such drugs on exacerbation duration.
The RESTORE (Reducing Exacerbations and Symptoms by Treatment with ORal Erdosteine in COPD) study was a prospective randomised, double-blind, placebo-controlled study, enrolling patients aged 40–80 years with Global Initiative for Chronic Obstructive Lung Disease stage II/III. Patients received erdosteine 300 mg twice daily or placebo added to usual COPD therapy for 12 months. The primary outcome was the number of acute exacerbations during the study.
In the pre-specified intention-to-treat population of 445 patients (74% male; mean age 64.8 years, forced expiratory volume in 1 s 51.8% predicted) erdosteine reduced the exacerbation rate by 19.4% (0.91 versus. 1.13 exacerbations·patient−1·year−1 for erdosteine and placebo, respectively; p=0.01), due to an effect on mild events; the reduction in the rate of mild exacerbations was 57.1% (0.23 versus 0.54 exacerbations·patient−1·year−1 for erdosteine and placebo, respectively; p=0.002). No significant difference was observed in the rate of moderate and severe exacerbations (0.68 versus 0.59 exacerbations·patient−1·year−1 for erdosteine and placebo, respectively; p=0.054) despite a trend in favour of the comparison group. Erdosteine decreased the exacerbation duration irrespective of event severity by 24.6% (9.55 versus 12.63 days for erdosteine and placebo, respectively; p=0.023). Erdosteine significantly improved subject and physician subjective severity scores (p=0.022 and p=0.048, respectively), and reduced the use of reliever medication (p<0.001), but did not affect the St George's Respiratory Questionnaire score or the time to first exacerbation.
In patients with COPD, erdosteine can reduce both the rate and duration of exacerbations. The percentage of patients with adverse events was similar in both the placebo and erdosteine treatment groups.
Abstract
RESTORE study: erdosteine reduces both rate and duration of COPD exacerbations with a placebo-like safety profile http://ow.ly/BbGI30dRdEt
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide, and imposes a considerable burden on healthcare resources. The current goals of COPD management are to increase the time free of disease symptoms, reduce time spent in hospital, and improve exercise tolerance, health status and quality of life (QoL) [1]. Exacerbations are an important area of COPD care due to their impact on health status and disease progression [2–5]. Current pharmacological treatment reduces the frequency of exacerbations, but does not abolish them [2, 6, 7]. Recent data indicate that the burden of COPD is also related to the duration of exacerbation, with the length of acute exacerbations being strongly linked to morbidity, poorer health status and hospital admissions. Moreover, longer exacerbations are associated with an increased risk of a new event and accelerate the rate of decline in lung function [8–10].
Oxidative stress, inflammation and mucus hypersecretion are among the factors driving the pathophysiology of COPD exacerbations. The exacerbation rate can be reduced by drugs with antioxidant, anti-inflammatory and mucolytic properties [11, 12]. However, no data are available on their efficacy in reducing the duration of exacerbations.
Erdosteine is a more recently developed mucoactive drug which addresses some of the problems associated with older drugs in this class [13]. Erdosteine has antioxidant and anti-inflammatory properties, but also modulates bacterial adhesiveness, properties potentially of value in the treatment or prevention of exacerbations [14–16]. Preliminary clinical data (EQUALIFE study) suggested that erdosteine, as an add-on therapy, could reduce the exacerbation rate and time spent in hospital, and improve health-related QoL in COPD patients [17]. However, the duration of the treatment (8 months over the winter period) and the size of that study were insufficient to assess the suitability of erdosteine as maintenance therapy in outpatients with COPD at risk of exacerbations treated with conventional COPD therapy.
In this study we assessed whether long-term treatment with erdosteine at its standard dose could reduce not only the exacerbation rate, but also impact the duration of exacerbations and time to next exacerbation event.
Methods
Study design
The RESTORE (Reducing Exacerbations and Symptoms by Treatment with ORal Erdosteine in COPD) study was a phase III multinational, randomised, double-blind, placebo-controlled parallel group study to evaluate the efficacy and safety of erdosteine added to usual maintenance therapy versus placebo over 12 months, a period long enough to avoid bias due to seasonal variability in exacerbation frequency. Patients with stable moderate-to-severe COPD were enrolled at 47 hospital-based pulmonary clinics in 10 European countries (see details in the supplementary material).
The study was conducted according to the International Conference on Harmonisation's E6 Good Clinical Practice Consolidated Guidance, Directive 2001/20/EU and the Declaration of Helsinki. The study was approved by local independent ethics committees.
Patients
Eligible patients were current or ex-smokers (≥10 pack-years) aged 40–80 years treated as outpatients, Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage II/III with a stable therapeutic regimen for ≥8 weeks. Patients had to have experienced two or more acute COPD exacerbations requiring medical intervention in the previous 12 months, but no exacerbations in the previous 2 months, with forced expiratory volume in 1 s (FEV1) 30–70% predicted and post-bronchodilator FEV1/forced vital capacity (FVC) <70% (the full inclusion and exclusion criteria are described in the supplementary material). Written informed consent was obtained from all patients prior to enrolment.
Randomisation and masking
Patients were randomised following their assigned number after their 2-week run-in period to receive either oral erdosteine at the approved dosage of 300 mg twice daily or placebo in addition to their usual maintenance therapy for COPD based on local physician practice and the available guidelines on COPD.
If respiratory symptoms worsened, patients were allowed to use short-acting β2-agonists as “reliever medications”, adding them to their usual prescribed maintenance therapy and to their trial treatment. This use was recorded in the patient diaries on a daily basis and further confirmed by the investigator in the case report forms. After randomisation, clinical visits occurred at 1, 3, 6, 9 and 12 months. No follow-up study was planned.
We stratified patients 1:1 according to concomitant inhaled corticosteroid (ICS) use in order to avoid any imbalance in the study treatment groups. The subgroups consisted of patients using bronchodilators alone and those using long-acting β2-agonists (LABAs) plus ICSs. An independent statistician generated a randomisation list of patient random numbers using a pseudo-random number generator. Series of four patients for each of the two strata were assigned to study sites in order to achieve, within each centre, a balanced number of patients treated with erdosteine or placebo in each of the two strata.
Erdosteine and placebo capsules were manufactured and provided by the sponsor (Edmond Pharma, Milan, Italy). The placebo was identical in composition, shape, colour and size, but did not contain any active ingredients. Erdosteine or placebo capsules were packed identically.
The investigator or anyone at the study site was prevented from knowing the allocation sequence with code labelling. The sponsor and clinical research associate were notified if there was a clinical reason for an individual's treatment to be unmasked by the investigator.
Outcomes
The primary efficacy outcome was the number of acute exacerbations occurring over the 12-month study period. Key secondary outcomes were the duration of exacerbations and time to first exacerbation.
At the time when the study was designed our original statistician advised that a simple comparison of the number of events in each group was sufficient. After the trial started it became clear that the most appropriate methodology was to use a modelled approach to be consistent with current statistical practice [18].
Exacerbations were defined as a symptomatic worsening beyond normal day-to-day variations and requiring a change in regular medication and/or healthcare resources utilisation: mild (increased use of bronchodilators), moderate (treatment with antibiotics and/or systemic corticosteroids and additional medical assistance) and severe (visit to an emergency department/hospitalisation) [19].
Exacerbations were collected using the patient's home daily diaries (see supplementary material). In these diaries, patients scored each of the following COPD symptoms on a five-point scale: difficulty in breathing, cough and trouble with sputum. Additionally, patients recorded any change in use of regular medication. Patients answered specific questions on the state of their disease (judgement about worsening of symptoms, extra medical visits and prescription/use of additional drugs for COPD treatment) on a weekly basis.
The investigators identified exacerbations by daily symptom score variation, change in regular medication and use of additional medication or an emergency hospitalisation for COPD. The onset of a COPD exacerbation was defined as an event with at least 2 days of symptomatic worsening and/or an increased level of healthcare utilisation.
The end of each event was established by a return to the pre-event symptomatology in the case of mild events and, by similar criteria, the cessation of additional treatment to the background therapy in the case of moderate events or severe events, whichever was longer. Investigators registered a new exacerbation provided there was an interval of at least 10 days without symptom variation and/or additional drug intake, and/or a re-hospitalisation since a previous episode.
Additional outcomes included subject and physician assessment of disease severity using a four-point numeric graded scale, where subjects and physicians rated from 0 to 4 how troublesome their lung problems were and how physicians rate patient's COPD symptoms (this scoring scale has not been formally validated), and the St George's Respiratory Questionnaire (SGRQ), hospitalisation rate, morning pre-dose FEV1, FVC and use of reliever medication, plus the 6-min walk test (6MWT; described in the supplementary material). Safety parameters monitored adverse events, study withdrawal rate, changes in vital signs, ECG parameters (including corrected QT) and routine laboratory testing.
Statistical analysis
We based the sample size for RESTORE on studies of mucoactive drugs available at the time of protocol design [20]. The sample size was calculated using the Andersen–Gill multiplicative intensity model for comparison of two treatment groups. The following assumptions were made: two-tailed test, exacerbations percentage reduction 20, average number of exacerbations 1.3 patient−1, type I error 0.05, type II error 0.20. As a result, a study population of 470 patients was needed.
Efficacy and safety analyses were conducted on all randomised patients who received at least one administration of the study treatment, with at least one available post-baseline efficacy evaluation.
A predefined analysis of exacerbation frequency was performed using a Poisson mixture regression model with adjustment for over-dispersion with the covariates of treatment, age, sex, body mass index and FEV1 at baseline to estimate the rate ratio with 95% confidence intervals [18]. A rate ratio <1 indicates a protective effect of treatment. All data were analysed per intention to treat. The natural logarithm of the duration, expressed as months in the study, was used as an offset variable to correct for differences in the time individuals spent under observation. A mixed regression model was used to correct for over-dispersion. Rate ratios from this model are expressed as percentages of reduction. Repeating the analysis according to the use of ICSs at baseline demonstrated no significant differences in exacerbation rate or duration between patients using ICSs or those not using them in both the placebo and erdosteine groups. Time to first exacerbation was evaluated by the log-rank test and described by the Kaplan–Meier method.
As per protocol, the secondary outcomes were compared by nonparametric statistics. After normality testing (Kolmogorov–Smirnov test) and eventual log-transformation for normalisation purposes, pulmonary function parameters, QoL scores and distance (metres) walked in the 6MWT were analysed using an ANCOVA model with treatment and centre (country) as variants, and baseline as a covariate. For use of “reliever” medications, the median data during the 12-month study period were analysed using the Wilcoxon rank-sum test. The proportion of patients presenting adverse events, adverse events leading to withdrawal, adverse drug reactions and serious adverse events were evaluated for each treatment group. We coded adverse events according to the Medical Dictionary for Regulatory Activities (www.meddra.org). The proportion of patients presenting adverse events, those adverse events leading to withdrawal, adverse drug reactions and serious adverse events were tabulated for each treatment group (see supplementary material).
The distribution over time of the clinical laboratory parameters was analysed by describing and comparing frequencies of abnormal values at baseline and after 12 months of treatment by means of the McNemar test.
Comparisons within treatment were performed by calculating the 95% confidence intervals of the changes from baseline, when applicable. All statistical tests were performed using SPSS version 21.0 (IBM, Armonk, NY, USA). A two-sided p-value <0.05 was considered significant for all tests.
Role of the funding source
All authors (academic investigators (R.W.D.N., J.A.W., M.I., G.F., C.P., A.F.C. and P.M.A.C.) and employee of the sponsor (E.P.)) had full access to and interpreted the data, and were responsible for the decision to publish the article. The sponsor did not place any restrictions on the academic authors for the decision to submit the findings for publication.
Results
Between November 2, 2010 and February 3, 2014, 467 patients were randomised and treated (528 patients screened). The initial sample size target was 470 and therefore the risk of false discovery is small.
Enrolment was slow due to the stringent inclusion criteria and was closed in 2014.
Figure 1 shows the trial profile of the study. Reasons for screening failure were similar in each treatment group. Enrolled patients were current smokers (n=135 (29%)) or ex-smokers (n=332 (71%)). There was no statistical difference between treatment groups in mean number of exacerbations during the 12 months before treatment (2.39 versus 2.30 exacerbations for erdosteine and placebo, respectively).
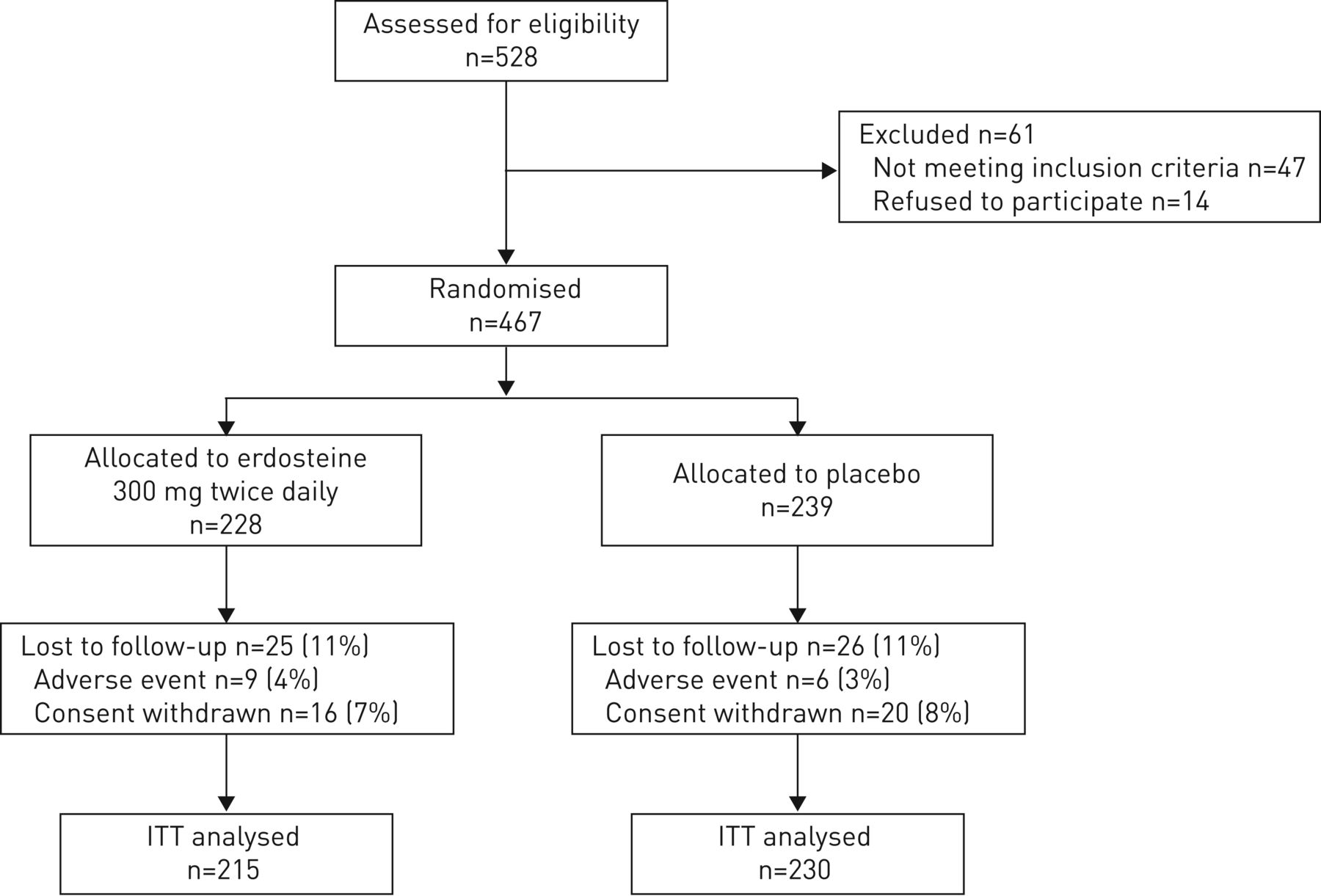
Trial profile. Percentages are based on the number of randomised patients in the single treatment group. ITT: intention to treat.
Baseline characteristics, including allowed medications for COPD treatment, were similar in the two treatment groups (table 1). Mean age was higher in subjects who exacerbated than in those who did not (p<0.05), although there were no significant differences between treatment groups. Mean post-bronchodilator FEV1 % pred was 52%. Disease severity was classified as GOLD stage II (moderate) in about half of the patients and GOLD stage III (severe) in most of the remaining patients. 60% of patients were taking a long-acting inhaled antimuscarinic drug, while 44% used a LABA plus ICS combination treatment.
Demographics and baseline characteristics
Baseline characteristics and lung function impairment were similar in patients who completed and those who did not complete treatment (table 2). Mean±sd treatment duration was 317.0±102.3 days in the erdosteine group and 318.1±105.2 days in the placebo group.
Baseline characteristics of patients who completed or did not completed the trial
During the trial, nine patients in the erdosteine group and six patients in the placebo group dropped out because of adverse events; 16 patients in the erdosteine group and 20 patients in the placebo group withdrew their consent to continue the study for other reasons. The dropout rate in both groups was similar at each time-point and at the end of the study (figure 1). Only one serious event in both groups was considered as drug related (one atrial fibrillation in the erdosteine group and one gall bladder empyema in the placebo group); two and four nonserious events in the erdosteine and placebo group, respectively, were considered as treatment related (see supplementary material).
457 exacerbations were recorded after 1 year of treatment: 196 (42.9%) exacerbations in the erdosteine group and 261 (57.1%) in the placebo group. 91 patients treated with erdosteine (42%) and 70 patients treated with placebo (30%) did not experience any exacerbation during the trial, with a relative risk of 0.8291 (p=0.01) in favour of erdosteine.
There was a 19.4% reduction in the exacerbation rate with erdosteine treatment (0.91 versus 1.13 exacerbations·patient−1·year−1 for erdosteine and placebo, respectively; p=0.01) as derived from the Poisson model (table 3).
Analysis of exacerbations
This result was driven by the reduction in the rate of mild exacerbations equal to 57.1% (0.23 versus 0.54 exacerbations·patient−1·year−1 for erdosteine and placebo, respectively, rate ratio 0.429; p=0.002). No statistically significant difference between the two patient groups was observed in the rates of moderate and severe exacerbations (0.68 versus 0.59 exacerbations·patient−1·year−1 for erdosteine and placebo, respectively, rate ratio 1.153; p=0.054) despite a trend in favour of the comparison group (figure 2).

Mean exacerbation rate in the study period (1 year). ns: not significant.
Erdosteine treatment was associated with a 24.6% decrease in the duration of all exacerbations (9.5±7.2 days with erdosteine versus 12.6±9.7 days with placebo; p=0.023). There was a positive effect of erdosteine on the duration of exacerbations both for mild and moderate/severe exacerbations patient groups (8.1±5.2 days with erdosteine versus 10.4±8.2 days with placebo; p=0.039 and 11.1±8.9 days with erdosteine versus 14.1±10.8 days with placebo; p=0.041, respectively) (figure 3). No significant differences were registered for both the exacerbation rate and duration among those patients taking ICSs and those patients not taking ICSs.

Mean exacerbation duration in the study period (1 year).
Although with a positive trend with erdosteine, the time to first exacerbation did not reach statistical significance (Kaplan–Meier plot of probability, p=0.07) (figure 4). Hospitalisations due to COPD exacerbations in the study were infrequent: 5.7% of the erdosteine-treated patients and 7.2% of the patients treated with placebo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier plot of probability of being exacerbation-free at each point through the study. Numbers show remaining patients at randomisation and during treatment at 50, 100, 150, 200, 250, 300 and 350 days.
Patient assessment of disease severity was significantly lower in the erdosteine group compared with the placebo group (1.48±0.74 versus 1.65±0.74, mean difference= −0.167; p=0.022). Similarly, physician global assessment of disease severity was significantly lower in the erdosteine group compared with the placebo group (1.53±0.73 versus 1.68±0.80, mean difference= −0.149; p=0.048). In the placebo group the SGRQ total score changed from 52.9±16.9 to 37.4±17.5 (p<0.001), while in the erdosteine group the SGRQ total score changed from 51.9±17.1 to 39.9±17.7 (p<0.001), without a significant difference between groups.
Only 10.2% of erdosteine-treated patients versus 33.7% of patients treated with placebo needed to increase their use of reliever medication during the trial (Fisher's exact test, p<0.001). There were no differences between active treatment and placebo in FEV1, FVC and 6MWT distance.
There were 706 adverse events reported during the study, 313 of them in the erdosteine group and 393 in the placebo group; only three (one serious) in the erdosteine group and five (one serious) in the placebo group were considered treatment related. There were 82 serious adverse events in 74 patients that occurred during the study, three of them (two in the placebo group and one in the erdosteine group) led to patient death (see supplementary material).
Discussion
Current pharmacological treatment of COPD improves, but does not abolish, exacerbation events. In RESTORE, erdosteine, a mucoactive drug added to usual COPD maintenance therapy, reduced the absolute number of exacerbations together with the event rate and their duration over 1 year. Both the patients and their physicians noted clinical improvements with active treatment, and less rescue medication was taken in this group. However, there was no significant effect on lung function, time to first exacerbations or 6MWT distance, nor was there a significant difference between groups in overall health status as measured by the SGRQ. These data have implications for the way we approach the treatment of COPD exacerbations and the role of erdosteine in their treatment.
The RESTORE trial provides further evidence that treatment with oral mucoactive drugs with antioxidant properties can reduce the frequency of COPD exacerbations. The primary analysis is statistically significant even if the effect size was smaller than expected, the event rate lower and we had fewer people recruited than those requested by the power calculation. This probably reflects the fact that our original estimates were very prudent and we assumed a higher dropout rate from the trial than we finally observed. There are other recent studies where the exacerbation rate was substantially lower than anticipated [21].
As with studies conducted in Asia with other mucolytic drugs, the total event rate was reduced but the time to the first event was not [11, 12]. Similar findings were reported in the initial trials of ICSs in COPD [22], but not when larger studies were reported [7, 23], and so this negative finding may be a function of our study size.
The reduction in the number of exacerbations we registered was driven largely by a reduction in the number of symptomatic events managed by an increase in bronchodilator use. In clinical practice, these mild symptomatic episodes are frequently seen especially in less advanced COPD stages, and they are usually unreported and consequently untreated with antibiotics and/or corticosteroids [24]. Recent data suggest that these mild events are associated with more healthcare use and worse outcomes, although the economic impact of mild exacerbations alone is likely to be less than moderate or severe events [25]. The ability of erdosteine to prevent these milder events may improve adverse outcomes associated with exacerbations and merits study over a longer timescale.
RESTORE was conducted in European patients with a higher background use of ICSs than in earlier trials with mucoactive drugs. Unlike the BRONCUS study where N-acetylcysteine did not reduce exacerbations in ICS-treated patients, we saw no influence of ICS use on the efficacy of erdosteine [20]. This may reflect the additional effect of erdosteine on bacterial adhesion, but may also be due to the inclusion in our exacerbation definition of some events identified by symptomatic change rather than the use of systemic corticosteroids [16]. These events are associated with impaired well-being even in relatively mild COPD [26].
Differences in the speed of onset and duration of exacerbations have been reported from detailed analysis of home diary card data, with a significant number of events beginning slowly and taking longer to resolve [8, 27]. The RESTORE study showed that erdosteine reduced the duration of all types of exacerbation, thus providing a longer time free from symptoms between events and reducing the total burden of these events on the patient. Like other drugs in this class, erdosteine had no effect on lung function or walking distance. This reflects the known biological properties of this drug, and suggests that the effects on exacerbation rate and duration were not due to altered lung mechanics.
There was no difference in respiratory health status as measured by the SGRQ between treatments, although like many other clinical trials there was a substantial improvement in SGRQ score in both treatment groups which may have dwarfed a modest change due to reduced exacerbation prevention. In contrast, the use of reliever treatment fell significantly with erdosteine treatment. Changes in reliever medication use have been seen previously with studies of LABA plus ICS combinations and are usually attributed to the use of a maintenance bronchodilator drug [28]. This cannot be the explanation here given the lack of impact of erdosteine on lung function, which in any case is not a bronchodilator. Reliever use is a risk factor for subsequent exacerbation, and its reduction with erdosteine is in keeping with the overall physician and patient statements that the treatment was significantly more effective than placebo [29]. The freedom from adverse events in the erdosteine group did mean that treatment was well tolerated and likely contributed to the low withdrawal rate from the study.
There are both strengths and limitations to our data. This study was designed and implemented before data from more recent studies were available, and so this study was powered using data from the earlier BRONCUS trial [11, 20]. The EXACT-PRO methodology to detect milder exacerbations had not been reported when RESTORE began so we have relied on daily diary cards scored by the investigator blind to treatment allocation to define exacerbations [30]. Our method for the detection of the duration of the events was pragmatic, and used information from both diary cards and healthcare utilisation to determine when events began and ended. As expected, milder events were shorter than more severe events and a treatment effect was observed in both. The blinded nature of this evaluation and the study size give us confidence that these observations are likely to be valid.
We elected not to include patients with very severe COPD in the study and further work is needed to determine whether erdosteine might also be beneficial in this population. Inevitably, the background maintenance therapy used reflects the choices available when the study was conducted and does not take account of more recent treatment approaches, such as dual long-acting bronchodilator treatment [31]. Nonetheless, our patients used more background treatment and specifically ICSs than was the case in earlier studies with mucoactive drugs. There was no bias due to differential withdrawal from the placebo arm or loss of study participants due to unacceptable side-effects [32]. Although the rate of moderate/severe exacerbations was lower than anticipated, there were still significantly more events than reported in recent larger studies where treatment differences were identified [21].
In conclusion, our data provide evidence that mucoactive drugs, like erdosteine, prevent exacerbations and shorten their duration irrespective of whether ICSs are given. However, the availability of a well-tolerated simply administered orally active treatment like erdosteine should be of value to patients who experience these troublesome episodes.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00711-2017_Supplement
Disclosures
Supplementary Material
P.M.A. Calverley ERJ-00711-2017_Calverley
A.F. Cicero ERJ-00711-2017_Cicero
R.W. Dal Negro ERJ-00711-2017_Dal_Negro
G. Fontana ERJ-00711-2017_Fontana
M. Iversen ERJ-00711-2017_Iversen
C. Page ERJ-00711-2017_Page
E. Pozzi ERJ-00711-2017_Pozzi
J.A. Wedzicha ERJ-00711-2017_Wedzicha
Acknowledgements
The authors thank all the participants and study investigators involved in the RESTORE study. Belgium: M. Decramer; Bulgaria: D. Osmanliev, K. Kostov, M. Peneva; Czech Republic: V. Kolek, V. Kasak, I. Pavlisova, J. Mares, P. Kalina, M. Buresova, D. Velart, R. Ivankova, R. Hrdina; Denmark: P. Lange; France: M. Aubier, J.F. Muir; Italy: S. Tognella, C. Balestriero, G. Scarmagnan, F. Zecchinato, S. Amaducci, M. Villani, S. Scoditti, F. Lavorini, M. Moretti, A. Tubaldi, G. Mattioli, E. Guffanti, A. Fumagalli, M. Del Donno, S. Centanni; Poland: J. Milanowski, H. Batura-Gabryel, M. Gabricki, E. Nizankowska-Mogilnicka, R. Chazan, J. Klimiuk, I. Grzelewska-Rzymowska, S. From, Z. Bartuzy; Romania: V. Tudorache, M. Marc, I. Iovan, D. Vancea, F. Mihaltan, R. Ulmeanu, T. Mihaescu, G. Jimborean, D. Todea, M.C. Pop, D. Bumbacea; Slovakia: H. Lescisinova, I. Jonner, A. Golubov; UK: A. Morice, L. Davies, L. McGarvey.
Footnotes
This article has supplementary material available from erj.ersjournals.com
The study is registered at EudraCT with identifier number 2008-008192-34 and at ClinicalTrials.gov with identifier number NCT01032304.
Support statement: Funding, medications and investigator's meeting costs for this study were provided by Edmond Pharma. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received April 5, 2017.
- Accepted July 15, 2017.
- Copyright ©ERS 2017
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










