





Abstract
Asthma is a chronic respiratory condition frequently associated with aberrant airway and systemic inflammation. Various inflammatory phenotypes in asthmatic airways have been described that relate to clinical phenotypes and impact on responses to conventional and novel asthma therapies. Macrophages are abundant immunocytes in the lung, capable of mounting diverse responses required for homeostasis and defence against pathogens.
Here, we summarise the clinical evidence regarding macrophage dysfunction in asthma. We also describe evidence supporting the role of macrophages as therapeutic targets in asthma. We conclude that macrophage dysfunction in asthma is highly prevalent and heterogeneous, and hypothesise that macrophages may play roles in promoting the discrete inflammatory phenotypes of asthma.
These clinical findings, along with recent ground-breaking insights into the ontogeny, behavioural complexity and longevity of pulmonary macrophages, support continued research into the role of macrophages as disease modifiers, biomarkers and therapeutic targets in asthma.
Abstract
Lung macrophages display numerous, therapeutically targetable, context-specific dysfunctions in asthma http://ow.ly/g14430cQhp4
Asthma and airway inflammation
Asthma is a heterogeneous disease characterised by respiratory symptoms including shortness of breath, wheeze and chest tightness, which result from variable airflow limitation. Chronic airway inflammation is common and heterogeneous in asthma, and its characterisation is central to the development of effective and efficient personalised programmes of asthma care. In relation to this, airway inflammation has been proposed as one of the core “treatable traits” in chronic respiratory disease [1, 2]. The emergence of targeted biological therapies has further stimulated research into asthma inflammatory phenotypes, which has proven critical for the success of recent clinical trials and in the identification of patients most likely to benefit from these relatively costly new treatments in the clinic [3].
Characterisation of airway inflammation has centred on identification and enumeration of discrete immune cell populations in the upper and lower airways [4]. Based on this quantification, patients may be classed into one of four inflammatory phenotypes of asthma: eosinophilic (high eosinophils, normal neutrophils), neutrophilic (normal eosinophils, high neutrophils), mixed granulocytic (high eosinophils, high neutrophils) and paucigranulocytic (normal eosinophils, normal neutrophils) [5]. While this system of phenotyping of asthma is of particular relevance in the context of this review, the burgeoning complexity of asthma phenotyping (based on clinical features and biomarkers) should be noted (for review, see [6, 7]). Importantly these inflammatory phenotypes have value in predicting patient clinical phenotype and in guidance of pharmacological therapy [6, 8]. In addition to eosinophils and neutrophils, numerous other cell types are present in asthmatic airways, including lymphocytes, mast cells and epithelial cells. The most abundant cells are often macrophages and monocytes, whose numbers remain relatively consistent between patients and across disease phenotypes [5].
Perhaps due to this limited fluctuation in macrophage numbers, there has been limited research into their potential role in asthma pathogenesis, as biomarkers and as targets for asthma therapy. Here, we summarise the existing research on macrophages in asthma from clinical studies, to examine their role as potential drug targets and in disease pathogenesis. We propose that rather than acting as passive bystanders to disease, diverse macrophage function/dysfunction may contribute to the varying inflammatory phenotypes of asthma.
Macrophage function and ontogeny
Macrophages are innate immune cells that possess numerous and diverse functions relating to their role as initial responders to immune stimuli and the development of the ensuing innate and adaptive immune response (figure 1). These functions include recognition, response to and destruction of invading microorganisms, antigen presentation, maintenance of tissue and immune homeostasis through removal of cellular debris, and production of inflammation modulating products. In addition, upon recognition of inflammatory stimuli, macrophages are capable of mounting powerful (and potentially damaging) inflammatory responses.
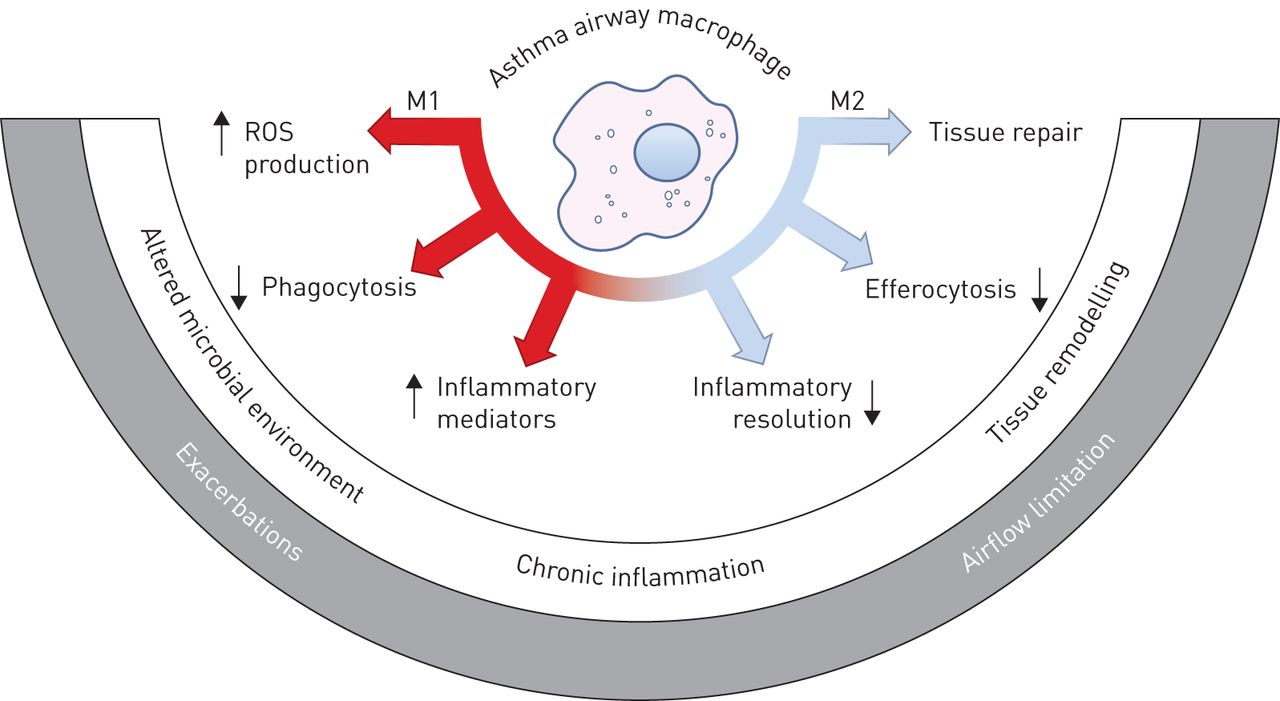
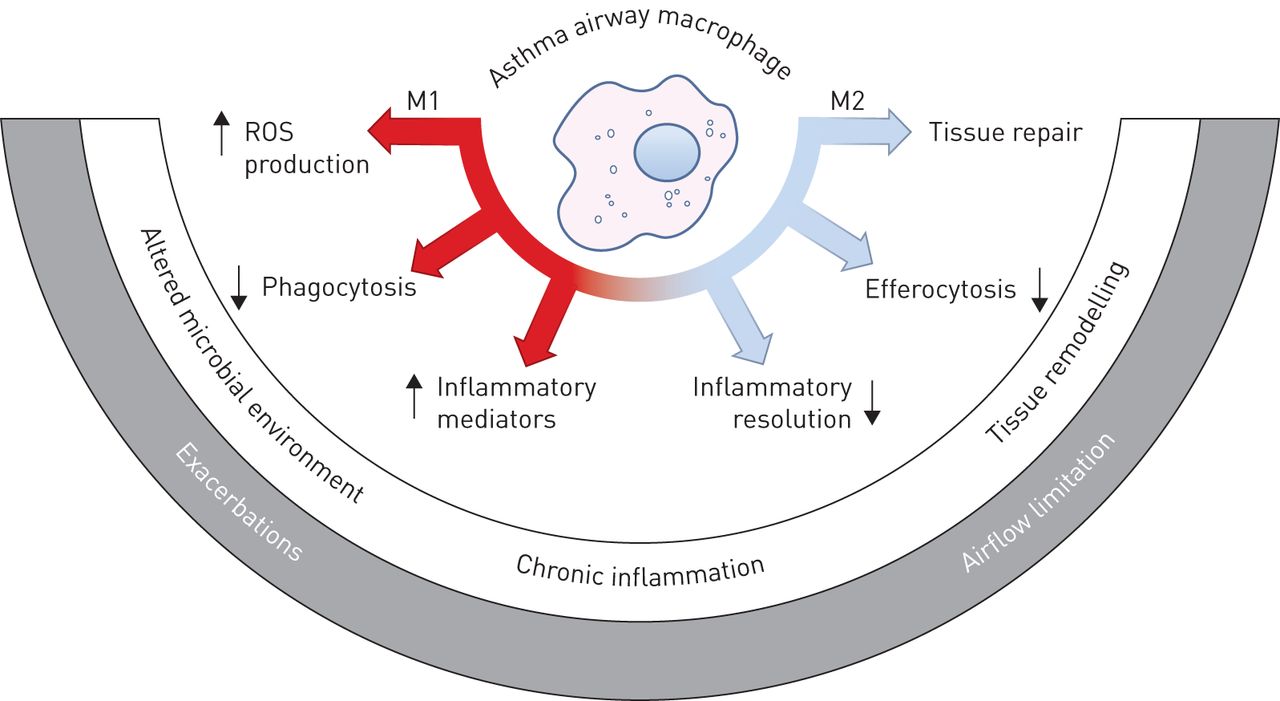
Macrophage dysfunction in asthma. Macrophages exert a variety of pro- and anti-inflammatory functions that are correlated with various states of immune activation and are broadly classed into M1- or M2-related classes. These important immune effector and homeostatic macrophage functions are altered in the asthmatic lung, and could contribute to and underpin discrete asthma pathologies and phenotypes. ROS: reactive oxygen species.
The lung contains at least two distinct resident macrophage populations, and recent studies have informed a paradigm shift in our understanding of the ontogeny, maintenance and activation of discrete lung macrophage populations [9–11]. Alveolar macrophages reside in the airway lumen and are a relatively long-lived population. Alveolar macrophages were initially believed to be seeded and continually replenished from a pool of circulating monocytes; however, extensive cellular fate mapping in mice has revealed that the during fetal development the lung is first seeded with yolk-sac-derived macrophages, followed by fetal monocytes, the latter of which differentiate into and account for the majority of alveolar macrophages in the adult. In adulthood, the alveolar macrophages pool is maintained by proliferation in situ [12–15]. The literature surrounding interstitial macrophage longevity and origin is less clear, with evidence supporting both yolk-sac- and bone-marrow-derived interstitial macrophage populations, at least in mice (for review, see [10, 16]). Like alveolar macrophages, interstitial macrophages appear capable of self-renewal through resident lung populations independently of infiltrating bone-marrow-derived monocytes. Little is known regarding macrophages residing in the bronchial lumen, and it remains unclear whether airway macrophages (isolated from bronchi using induced sputum) and alveolar macrophages (from bronchoalveolar lavage (BAL)) share a common ontogeny and function. The lung is also host to a resident monocyte population about which very little is known, other than they do not differentiate into alveolar macrophages and interstitial macrophages under normal conditions [9].
A further layer of complexity in the adult lung arises during inflammation and subsequent resolution of inflammation, as circulating monocytes are recruited to lung tissue and airways. These infiltrating monocytes possess unique functional roles and are also capable of differentiating under tissue-specific cues into alveolar macrophages that mimic resident embryonic-derived alveolar macrophage populations from a functional perspective [11]. Detailed transcriptomic analyses of bone-marrow-derived alveolar macrophages reveals differences in relatively few transcripts when compared with resident embryonic-derived alveolar macrophage populations, suggesting that although macrophage ontogeny contributes to macrophage phenotype within unique tissue microenvironments, tissue environmental-specific cues appear to play a dominant role [11, 17]. Differing origin and tissue-specific cues likely result in distinct functional output of alveolar macrophages, interstitial macrophages and pulmonary monocytes, with alveolar macrophages proposed to play a homeostatic suppressive innate immune role [18–20].
The distinct functions of subtypes of interstitial macrophages are poorly understood, and further work is also required to understand the potentially pathogenic roles of infiltrating monocytes and monocyte-derived macrophages. Of note, the recent advances in our understanding of pulmonary macrophage ontogeny stem largely from mouse studies, and further work will be required to define appropriate markers and means of monitoring and understanding the role of the various lung macrophage/monocyte populations in the pathogenesis and treatment of asthma, in particular in clinical samples.
Macrophage activation
Macrophages have somewhat contradictory functional roles, with involvement in both pro- and anti-inflammatory processes. This behavioural diversity is largely dictated by ontogeny, tissue-derived cues (as discussed earlier) and exposure to ever-changing environmental stimuli. In general, pro-inflammatory innate immune functions have been ascribed to classically activated (or M1) macrophages, which arise following stimulation by pro-inflammatory molecules, including lipopolysaccharide (LPS), tumour necrosis factor (TNF)-α and interferon (IFN)-γ. In contrast, functions relating to parasite destruction, immune resolution and tissue remodelling are more pronounced in alternatively activated (or M2) macrophages, which can be elicited by type 2 cytokines (e.g. interleukin (IL)-4 and IL-13). This classification is largely based on in vitro studies and recent in vivo work has shown that macrophages, including alveolar macrophages, exhibit a spectrum of activation states far more complex than this simple M1/M2 classification [21, 22]. Importantly, macrophages are capable of switching between these established and contrasting activation states, at least in vitro [23].
The key features of pulmonary macrophage biology (longevity, functional diversity and plasticity) highlight the potential for macrophage involvement in disease pathogenesis, and their utility as both biomarkers and drug targets in chronic inflammatory respiratory conditions, including asthma.
Macrophage functions altered in asthma
Phagocytosis
Altered microbial colonisation of the airways is a feature of asthma. Inflammatory and clinical phenotypes of asthma are associated with distinct airway microbial signatures, although the causes and functional significance of airway dysbiosis in asthma remain unknown [24–28]. As abundant phagocytes, macrophages likely play a key role in shaping microbial diversity in the airways of asthmatic patients. Several clinical studies have established that macrophage phagocytosis is dysfunctional in asthma [22–30].
Defective phagocytosis of bacteria, yeast and carbon particulate matter has been demonstrated in alveolar macrophages and peripheral blood monocytes of asthmatic patients [22–30]. Severe asthma is associated with a defect in alveolar macrophage phagocytosis of the bacteria Haemophilus influenzae and Staphylococcus aureus, which are associated with airway dysbiosis in asthma [28, 29]. This defect in phagocytosis appears not to be limited to the airways, as monocyte-derived macrophages from severe asthmatic patients also exhibit deficient phagocytosis when compared with those derived from healthy and mild asthmatic patients. Treatment ex vivo with dexamethasone does not impair phagocytosis by monocyte-derived macrophages in any patient group, suggesting that defective phagocytosis observed in severe asthma alveolar macrophages is not due to high-dose corticosteroid treatment of these patients [30].
Similarly, in children, increasing asthma severity is associated with increased markers of oxidative stress and reduced bacterial phagocytosis in alveolar macrophages [31, 32]. Ex vivo supplementation with the antioxidant glutathione rescues alveolar macrophage phagocytosis, implicating oxidative stress as a potential contributor to impaired phagocytosis in severe asthma [31]. Alveolar macrophages from allergic asthmatic and aspirin-sensitive asthmatic patients display reduced phagocytosis of yeast zymosan bioparticles and production of associated effector molecules [33]. Phagocytosis of opsonised yeast by peripheral blood monocytes is impaired in asthmatic children, irrespective of asthma severity [34]. These defects in yeast bioparticle phagocytosis and downstream signalling are associated with the extent of BAL eosinophilia [33, 35]. The eicosanoid prostaglandin E2 (PGE2), which is elevated in the sputum of eosinophilic and severe asthma patients, has been linked to decreased phagocytosis of particulate carbon matter in airway macrophages of asthmatic children [36, 37].
To summarise, impaired macrophage phagocytosis of microbial and particulate matter is a feature of asthma. This impairment could contribute to the altered microbial environment and relate to bacterial-induced disease exacerbations, which characterise severe forms of asthma. Initial findings suggest that oxidant/antioxidant balance and the extracellular signalling milieu may play significant roles in the inhibition of macrophage phagocytosis in asthma. Importantly, these factors may be targeted for therapeutic gain.
Efferocytosis
Efferocytosis is the process by which dead, dying or stressed host cells are recognised, engulfed and digested by neighbouring cells. Macrophages are key mediators of efferocytosis, which plays a critical immune regulatory function. Rapid and efficient clearance of dying cells prevents or limits the release of damage-associated molecular patterns (DAMPs) and associated inflammation. Dead cells and DAMPs (e.g. high mobility group box 1) are elevated in asthmatic airways, consistent with a role for impaired efferocytosis in contributing to aberrant inflammation [38–40]. Importantly, efferocytosis is also a crucial mechanism for neutrophil turnover [41]. Thus, impaired efferocytosis could result in neutrophil persistence and contribute to the neutrophilic asthma phenotype. The efferocytotic capacity of macrophages of the upper and lower airways is reduced in asthma, particularly in individuals with noneosinophilic asthma [42–44].
Unique disease-related variables appear to have a significant impact on macrophage efferocytosis, with reduced efferocytosis observed in noneosinophilic versus eosinophilic asthma [42], obese versus nonobese asthma [43] and severe versus mild asthma [44]. It remains unclear whether impaired efferocytosis plays a role in promoting asthma pathology and symptoms. Further studies are required to elucidate the molecular mechanisms that underlie the efferocytosis defect in asthma, which could involve numerous receptors, bridging molecules and target molecules that mediate host cell recognition and uptake [41].
Inflammatory mediator production
In the airway lumen and bronchial mucosa, macrophages are major cellular sources of critical pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6 and IL-8 [45, 46]. Alveolar macrophages from asthmatic patients release more TNF-α, IL-1β, IL-6 and IL-8 than healthy controls, but display a dampened inflammatory response to LPS stimulation. In contrast to healthy alveolar macrophages, IL-4 treatment fails to significantly inhibit LPS-induced pro-inflammatory cytokine release in asthmatic alveolar macrophages [47–51]. The innate pro-inflammatory TNF-α and IL-1β signalling pathways are implicated in neutrophilic asthma [52]. This correlates with increased IL-1β production by airway macrophages in asthma [53], in particular neutrophilic asthma [54].
In addition to the airway lumen, macrophages and monocytes appear to be a significant source of increased TNF-α and IL-1β in asthmatic airway tissue and the bronchial epithelium [46, 55]. IL-17 is a key pro-inflammatory cytokine of the T-helper 17 (Th17) cell pathway and has been linked to a role in promoting neutrophilic airway inflammation in asthma [56]. Although Th17 cells are a major cellular source of IL-17, other immune cell types have been implicated in its production in asthma [57]. IL-17 is elevated in the BAL of asthmatic patients, which correlates with increased numbers of IL-17+ alveolar macrophages [58]. In addition, evidence suggests that alveolar macrophages play an important role in the late-phase inflammatory response of atopic asthmatic patients following allergen challenge, producing increased TNF-α and IL-6 [59].
The possible roles of macrophages in influencing varying adaptive immunity pathways in the asthmatic lung remain relatively unexplored. Of note, the enhanced production of IL-1β and IL-6 by asthmatic alveolar macrophages can drive increased IL-5 production by CD4+ T-cells, potentially enhancing eosinophilic inflammation in the airways of asthmatic patients [60].
To summarise, altered pro-inflammatory cytokine production by pulmonary macrophages likely contributes to chronic airway inflammation. Importantly, macrophages may adopt differing phenotypes in discrete inflammatory subsets of asthma.
Anti-inflammatory molecule production
IL-10 is a critical anti-inflammatory cytokine and is thought to be an important mediator of macrophage anti-inflammatory functions in the resolution of inflammation. IL-10 protein levels are decreased in the airways of asthmatic patients [61]. However, studies on airway macrophages have reached conflicting conclusions. While IL-10 mRNA is increased in alveolar macrophages and sputum macrophages [62–64], protein expression in macrophages is decreased [65–67], suggesting altered post-transcriptional or post-translational regulation of IL-10 in asthma alveolar macrophages. IL-12 expression is reduced and could contribute to the enhanced Th2-skewing activity displayed by alveolar macrophages from asthmatic patients versus healthy patients and associated enhanced IL-5 production from CD4+ T-cells [51, 68].
Transforming growth factor (TGF)-β is a pleiotropic cytokine produced by multiple cell types in the inflamed lung. TGF-β levels are increased in both the airways and mucosal tissue of asthmatic patients [69, 70]. Although TGF-β mRNA is increased in alveolar macrophages of asthmatic patients [64], considerable regulation occurs at the post-translational level and protein production by alveolar macrophages has not been explored. Mucosal expression appears largely limited to eosinophils, neutrophils and fibroblasts [70, 71].
Numerous studies have established that the balance between production of pro-inflammatory lipid mediators leukotrienes and anti-inflammatory lipoxins is disturbed in asthma. In particular, severe asthma in adults and children is associated with an increased leukotriene B4 (LTB4)/lipoxin A4 (LXA4) ratio, measurable both in the airways and on a systemic level [72–75]. Macrophages appear to be a critical cellular site of impaired lipoxin synthesis in severe asthma [72], which is associated with increased oxidative stress and can be reversed by inhibitors of soluble epoxide hydrolase [76]. How this balance of macrophage eicosanoid synthesis is perturbed in different inflammatory phenotypes of asthma is unknown at present; however, the measurement of the LTB4/LXA4 ratio in exhaled breath condensate may present a good opportunity for the development of a clinically useful biomarker [77].
Altered macrophage polarisation in asthma
Few clinical studies have addressed the relative abundance or role of M1 and M2 macrophages in asthma pathogenesis (reviewed by Jiang and Zhu [78]). Recent studies have indicated that M2 macrophages may be elevated in asthma based on marker molecule expression [79, 80]. The clinical studies described in the previous sections, however, indicate that a number of M1- and M2-related functions of macrophages are altered in asthma (figure 1). As recent studies indicate, macrophage activation and polarisation is immensely complex. Transcriptional analyses provide expanded frameworks to examine the reprogramming of airway macrophages in asthma [22, 81]. Given the plasticity and complexity of macrophage phenotypes in the lung in vivo, further comprehensive studies of macrophage polarisation in stable and exacerbating asthma would be of value.
Modulation of macrophage function by asthma therapies
Corticosteroids
Corticosteroids are one of the most commonly prescribed medications for the treatment of asthma. Corticosteroids exert anti-inflammatory effects and may target multiple cell types in asthma, including epithelial and immune cells [82], although macrophages are proposed to be one of the principle targets [83]. In asthma, corticosteroid stimulation of alveolar macrophages induces glucocorticoid receptor-α translocation to the nucleus and subsequent recruitment of histone deacetylases (HDACs) to target promoter sequences, often resulting in suppression of pro-inflammatory genes [83–85].
Effects of corticosteroids on macrophages in asthma
Numerous clinical trials have shown that treatment of asthmatic patients with corticosteroids can modulate airway macrophage functions (table 1). Corticosteroid treatment of asthma patients suppresses PGE2 and LTB4 production by alveolar macrophages [86–88]. A single dose of prednisone can suppress alveolar macrophage-mediated IL-1β production in the lower airways of asthmatic patients [53], and corticosteroids can reduce LPS-induced TNF-α, IL-1β, IL-6 and IL-8 to varying extents [84, 89, 90]. Corticosteroids may also function through enhancement of anti-inflammatory functions of alveolar macrophages, including IL-10 synthesis [66, 91]. Corticosteroids reduce oxidative stress in asthmatic airways through reduction of alveolar macrophage reactive oxygen species (ROS) formation [92]. Alveolar macrophage expression of the oxidative stress marker haem oxygenase-1 is increased in steroid-naive asthma and suppressed by corticosteroid treatment [93, 94].
Inflammatory outcomes in clinical trials of asthma therapeutics
Corticosteroid resistance in asthmatic macrophages
Responses to corticosteroids vary among asthma patients and individuals with severe asthma often respond poorly to corticosteroid treatment. Several clinical studies have shown that relative resistance of clinical features of asthma to corticosteroid treatment correlates with relative corticosteroid resistance of alveolar macrophages. This alveolar macrophage resistance manifests as reduced capacity of corticosteroids to suppress LPS-induced pro-inflammatory LTB4, TNF-α, IL-6, IL-1β and CXCL8 production [84, 95–97]. Alveolar macrophage inflammatory function is skewed towards an M1-like phenotype in corticosteroid-resistant asthmatic patients, which is associated with higher BAL endotoxin levels and altered microbial colonisation of the airways [27, 98].
Although a role for an altered airway microbiome has been proposed in the development of corticosteroid resistance in asthmatic alveolar macrophages [27], the finding that circulating monocytes of severe asthmatic patients also display corticosteroid resistance may challenge this hypothesis [99]. Corticosteroid resistance in asthmatic alveolar macrophages correlates with activation of the p38 signalling pathway [96] and p38 inhibitors can restore corticosteroid sensitivity to severe asthma alveolar macrophages ex vivo [95, 100]. Aberrant p38 pathway activity may drive corticosteroid resistance through phosphorylation and inhibition of glucocorticoid receptor-α, although upregulation of the inhibitory glucocorticoid receptor-β isoform in asthma alveolar macrophages may also play a role [84]. Of note, it has been suggested that the association of alveolar macrophage glucocorticoid insensitivity with clinical corticosteroid insensitivity could be used to guide pharmacotherapy [95].
To summarise, several clinical studies point towards the alveolar macrophages as a critical cellular site of corticosteroid resistance in severe forms of asthma.
Macrolides
Macrolide treatment has been associated with improved asthma outcomes for >40 years [101, 102]. Although used primarily as antibiotics, studies have established that macrolides possess additional immunomodulatory properties, a combination of which may render them particularly effective in asthma. Those asthma subtypes most likely to benefit from macrolide treatment include severe noneosinophilic asthma [103, 104], asthmatic patients that test positive for the presence of Mycoplasma pneumoniae and Chlamydia pneumoniae [105–107], and eosinophilic aspirin-sensitive asthma [108].
It remains unclear whether macrolides exert protective effects primarily through their antibacterial properties or whether immunomodulatory effects of macrolides on immune cells are required. The utility of this approach as a long-term therapy for asthma is an issue, given concerns about the emergence of bacterial resistance. The development of macrolides lacking antibiotic activity but retaining their immunomodulatory effects may help to address this [109, 110]. Of note, azithromycin treatment of both azithromycin-resistant and -susceptible pneumococci reduced TNF-α production in bacteria-exposed macrophage cells, providing evidence that antibacterial activities are not required for immunomodulation of macrophage function [111].
Effects of macrolides on macrophages
Clinical trials have confirmed immunomodulatory functions of macrolides in asthma, including reduction of cytokine expression (IL-8, TNF-α and IL-12) typically associated with macrophages (table 1) [103, 106]. Macrolides accumulate within the lysosomes of phagocytic cells, reaching concentrations far higher than those measured in extracellular compartments. In the lung, macrophages and neutrophils accumulate macrolides to the greatest extent and maintain high intracellular concentrations long after treatment, and thus could represent important cellular sites of immunomodulatory function in asthma [112–115].
Treatment of acutely exacerbating asthmatic children with clarithromycin reduced concentrations of macrophage-associated cytokines TNF-α, IL-1β and IL-10 in nasopharyngeal aspirates [116]. Macrolides can reduce airway IL-1β and TNF-α and alveolar macrophage IL-8 in patients with chronic lower respiratory tract infection, supporting suppression of M1 macrophage-related mediators as a mechanism of macrolide action in the lung [117]. Macrolide suppression of IL-8 production in alveolar macrophages was confirmed in a separate study [118]. These clinical studies are supported by numerous studies performed on isolated macrophages and macrophage cell lines in vitro [119].
Macrolide treatment also has impacts on macrophage phagocytosis and efferocytosis. Erythromycin and clarithromycin treatment enhance efferocytosis of apoptotic neutrophils by healthy human alveolar macrophages, independently of pro-inflammatory mediator production [120].
Although not studied in asthma, defective efferocytosis of alveolar macrophages from chronic obstructive pulmonary disease (COPD) patients was restored back to levels seen in healthy patients with azithromycin treatment both ex vivo and in vivo [121, 122]. In addition, azithromycin treatment of COPD patients improved bacterial phagocytosis by alveolar macrophages [123], although ex vivo azithromycin treatment only improved defective phagocytosis in monocyte-derived macrophages derived from smokers but not COPD patients [124]. Thus, macrolide treatment can rescue defective efferocytotic and phagocytic activity displayed by alveolar macrophages in COPD. As alveolar macrophages in severe and noneosinophilic asthmatic patients display similar defects in efferocytosis as those of COPD patients, it is plausible that macrolide treatment could exert similar effects in asthma. At present the mechanisms underlying macrolide-mediated rescue of defective alveolar macrophages efferocytosis are unknown [122].
Statins
Statins (5-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors) are widely prescribed cholesterol-lowering drugs. Statins have also been widely reported to exert immunomodulatory effects, including anti-inflammatory effects. Statin use is associated with reduced severe asthma exacerbation-related hospital and emergency department visits [125–127]. Given the link between airway inflammation and asthma exacerbation frequency [128], this suggests statins may at least in part act through anti-inflammatory mechanisms. Indeed, several clinical trials, while failing to show improvements in lung function, have demonstrated anti-inflammatory effects of statins in asthma (table 1).
Effects of statins on macrophages
Statin treatment in asthmatic patients enhances inhaled corticosteroid (ICS)-induced activation of the noncanonical NF-κB pathway in alveolar macrophages, resulting in enhanced indoleamine 2,3-dioxygenase transcription and enzymatic activity, which stimulates IL-10 production [129]. A separate clinical trial concluded that statin treatment reduces sputum macrophage counts and LTB4 levels [130]. Of note, a combination of ICS and atorvastatin treatment in smoking asthmatic patients significantly reduces sputum IL-1β, a cytokine produced by macrophages and neutrophils and linked to neutrophilic asthma phenotypes [131]. Thus, it is possible that statin treatment may benefit subsets of asthmatic patients with corticosteroid-insensitive inflammation, such as that associated with smoking and neutrophilic airway inflammation [132].
In alveolar macrophages isolated from COPD patients lovastatin restores defective efferocytosis in an HMG-CoA reductase-dependent manner [133]. As similar alveolar macrophage efferocytosis defects exist in severe and neutrophilic asthma, further clinical trials examining this potential therapeutic mechanism of statins are merited [42, 122]. Statins also exert anti-inflammatory effects in rhinovirus-infected asthmatic alveolar macrophages, implicating cholesterol biosynthesis in the macrophage antiviral response [134]. Atorvastatin inhibits the particulate-induced production of a number of critical innate immune mediators in alveolar macrophages, including IL-1β, granulocyte–macrophage colony-stimulating factor (GM-CSF), IL-6 and TNF-α. In contrast, pro-inflammatory cytokine production by primary bronchial epithelial cells induced by particulate matter is unaffected, supporting the role of macrophages as an important site of statin-mediated immunomodulation in the lung [135].
A recent large-scale screen of drugs in clinical use on macrophages identified statins as enhancing tetraspanin expression, which inhibits LPS-mediated inflammatory signalling through sequestration of the LPS signalling mediator CD14 [136]. This finding may have particular relevance in noneosinophilic asthma, which is associated with abnormal Toll-like receptor signalling and enhanced bacterial colonisation of the airways.
To summarise, mechanistic studies have demonstrated a number of mechanisms whereby statins suppress inflammation in lung macrophages, pointing towards promising avenues of research and highlighting potential asthmatic populations that may benefit most from statin treatment (e.g. frequent exacerbators, obese asthmatic patients, neutrophilic asthmatic patients and patients with airway dysbiosis).
Phosphodiesterase inhibitors
Theophylline
Theophylline has been used for the treatment of asthma for almost a century [137]. Theophylline was initially prescribed as a bronchodilator, although unfavourable side-effects associated with the doses required to achieve bronchodilation have seen it superseded and largely replaced by β-agonists. Theophylline exerts its biological effects through inhibition of phosphodiesterases (PDEs), including PDE-3 and PDE-4 isoforms, increasing intracellular concentrations of the second messenger molecules cAMP and cGMP. While PDE-3 inhibition is related to bronchodilatory effects, PDE-4 inhibition is strongly linked to immunomodulatory functions. Side-effects associated with theophylline, including nausea and headaches, also result from PDE inhibition.
Several clinical trials of theophylline in asthma have assessed the use of low doses, which maintain the anti-inflammatory effects while reducing side-effects (and bronchodilation) (table 1). These studies have demonstrated efficacy of low-dose theophylline as a steroid-sparing agent in mild-to-moderate asthma and beneficial effects in relatively corticosteroid-refractory patients who smoke or have severe asthma.
Effects of theophylline on macrophages
The corticosteroid-sparing effects of theophylline may involve induction of HDAC activity in alveolar macrophages [138]. Corticosteroids exert their anti-inflammatory effects, in part, by mediating recruitment of HDACs to pro-inflammatory gene promoter sequences, resulting in deacetylation and transcriptional suppression. Theophylline treatment of mild asthmatic patients increases HDAC activity and expression in bronchial biopsies [138]. In addition, alveolar macrophages from mild asthmatic patients display similar increases in HDAC activity in response to low-dose dexamethasone combined with theophylline versus high-dose dexamethasone alone. Theophylline alone is sufficient to increase HDAC activity, allowing greater suppression of inflammation when corticosteroids are added and synergistic inhibition of pro-inflammatory gene expression [85, 138].
Further studies have substantiated the role of macrophages as key targets for the immunomodulatory effects of theophylline in asthma. Alveolar macrophages isolated from mild asthmatic patients treated with theophylline have reduced pro-inflammatory LTB4 production and thus theophylline can reduce pro-inflammatory eicosanoid production by alveolar macrophages in vivo [139]. Theophylline also inhibits TNF-α and enhances IL-10 production in LPS-stimulated peripheral blood mononuclear cells isolated from asthmatic patients [140]. In addition, theophylline treatment of alveolar macrophages ex vivo inhibits zymosan-stimulated cAMP degradation and hydrogen peroxide generation [141].
While these studies demonstrated anti-inflammatory actions of theophylline on macrophages, most only reported effects following receipt of an exogenous pro-inflammatory stimulus. Of note, primary alveolar macrophages isolated directly from theophylline-treated mild asthmatic patients exhibit no change in baseline production of IL-10, GM-CSF or TNF-α, although eosinophil levels are reduced in the BAL of these patients [142]. Thus, while theophylline can inhibit baseline inflammatory characteristics of monocytes and alveolar macrophages when administered ex vivo [143, 144], it is likely that immunomodulatory targets additional to macrophages are also important in the clinic. A limitation of these studies is that they have largely reported on a limited range of potential pro-inflammatory outputs of macrophages. A transcriptomic approach revealed that theophylline exerts broad effects, highlighting the potential of theophylline to inhibit a number of alternative pro-inflammatory pathways, including the production of IL-13 [145]. Therefore, theophylline may inhibit both type 1 and type 2 cytokine production in alveolar macrophages, establishing a link to eosinophilic asthmatic subtypes that merits further exploration.
Roflumilast
Roflumilast is a potent selective PDE-4 inhibitor, which is thought to exert its anti-inflammatory action through inhibition of the PDE-4B isoform [146]. It is approved for the treatment of COPD and asthma. Clinical trials of roflumilast in asthma have established that it provides similar benefits to low/moderate-dose ICS treatment, assessed by improvement in lung function, asthma symptoms and reduced rescuer use (table 1) [147, 148]. In addition, several clinical trials have shown that short-term (up to 14 days of treatment) roflumilast dosing can inhibit both early and late asthmatic responses to allergen [149–151]. Roflumilast appears to be particularly effective at reducing the late asthmatic response, reducing allergen-induced sputum eosinophil and neutrophil influx, and associated inflammatory biomarkers in asthmatic individuals [151, 152].
Effects of roflumilast on macrophages
Macrophages express a number of PDE-4 isoforms, with PDE-4B being the principle target of roflumilast [153]. Roflumilast is anti-inflammatory in primary human lung macrophages, inhibiting LPS-stimulated release of pro-inflammatory chemokines and cytokines in a cyclooxygenase-2-dependent manner, via production of PGE2 [154]. Further studies are required to fully elucidate the critical cellular sites of anti-inflammatory roflumilast activity in asthma; however, based on current evidence, macrophages remain plausible candidate targets.
Leukotriene antagonists
The leukotriene signalling pathway is a target of several therapeutics used in maintenance therapy of asthma [155]. Leukotriene signalling is targeted in the clinic through use of cysteinyl leukotriene receptor antagonists (e.g. montelukast and zafirlukast) and inhibition of 5-lipoxygenase (zileuton), an enzyme involved in both cysteinyl leukotriene and LTB4 production. Antagonists of this pathway exert both bronchodilatory and anti-inflammatory effects.
Effects of leukotriene antagonism on macrophages
Alveolar macrophages both produce and respond to cysteinyl leukotrienes and LTB4, and the balance between these pro-inflammatory factors and anti-inflammatory mediators such as LXA4 is disturbed in asthma (as discussed earlier). In addition, monocytes, along with eosinophils, epithelial cells and airway smooth muscle cells, express cysteinyl leukotriene receptor.
Zileuton inhibits cysteinyl leukotriene and LTB4 production in stimulated alveolar macrophages as well as in other immunocytes, including eosinophils, neutrophils and mast cells [156–158]. Although it has been suggested that macrophages may indeed be an important therapeutic target of leukotriene antagonists [159], convincing clinical evidence is lacking.
Montelukast treatment of atopic asthmatic children reduces systemic TNF-α, which may be in part mediated by the inhibition of leukotriene signalling in monocytes [160]. The action of cysteinyl leukotriene on monocytes/macrophages results in increased migration, ROS production, release of matrix metalloproteinase-9 and production of pro-inflammatory cytokines [161]. Additionally, montelukast treatment of M2-polarised blood monocytes prevented LPS-induced upregulation of M2-associated cytokines, including IL-10 [162].
To summarise, leukotriene antagonists modulate numerous macrophage and monocyte functions relevant in the context of asthma. However, numerous cellular targets for these agents exist and further high-quality clinical evidence would be required to confirm a role of macrophages as therapeutic targets of leukotriene antagonists. Additional questions regarding the action of leukotriene antagonists on macrophages in asthma remain, such as whether the observed inhibitory effect on macrophage phagocytosis via the Fc receptor has clinical relevance [163] and whether variable macrophage phenotypes may underlie the differential response of atopic asthmatic patients to leukotriene antagonism in allergen challenge models [159].
β-Agonists
β-Agonists act as bronchodilators, acting principally to induce relaxation of airway smooth muscle. Short-acting β-agonists (SABAs) are a mainstay of asthma therapy, and are often prescribed as monotherapy in mild asthma or are used as reliever medications during periods of asthma exacerbation in mild and severe asthmatic patients [164]. Long-acting β-agonists (LABAs) are also widely prescribed as a therapeutic add-on to ICSs, the use of which largely negates the negative health impacts of LABAs [164, 165]. In addition to their bronchodilatory action on airway smooth muscle, β-agonists exert anti-inflammatory effects through numerous targets including eosinophils, T-lymphocytes, neutrophils and mast cells [166].
Effects of β-agonists on macrophages
Several studies have demonstrated the presence of β2-adrenergic receptors on alveolar macrophages [167] and their responsiveness to β-agonist stimulation [168, 169]. However, repeated use of β-agonists reduces surface β2-adrenergic receptor expression in lung macrophages and desensitises to further β-agonist stimulation [168, 170]. This phenomenon is also observed in other cell types, where it is reversible by concomitant treatment with corticosteroids. However, reversibility following corticosteroids treatment does not occur in alveolar macrophages [171]. Thus, lung macrophages in asthmatic patients on frequent SABA or LABA treatment are unlikely to respond strongly to β-agonist. Ex vivo stimulation of alveolar macrophages with opsonised zymosan or IgE/anti-IgE complexes induced inflammatory mediator production that was not inhibited by isoprenaline treatment. However, salbutamol treatment could suppress LTB4 production under the same conditions [172, 173].
To summarise, macrophages would not appear to be an important cellular target for bronchodilator and immunomodulator functions of β-agonists.
Conclusions and suggestion of knowledge gaps
Macrophages carry out a range of essential immune functions in the lungs and these functions are dysregulated in asthma. Preliminary evidence from clinical trials suggests that a number of macrophage functions may be targeted using conventional and novel treatments, and that targeting of macrophage (dys)function in asthma may form part of the therapeutic mechanism of action (figure 2). The description of hitherto underappreciated functional complexity and plasticity of macrophage phenotypes has highlighted a knowledge gap regarding the roles of macrophages in the pathogenesis of asthma. A few recent studies have begun to reveal that, much like asthma, macrophage dysfunction in asthma is heterogeneous, further strengthening the argument for their involvement as pivotal disease modulators.

{kind=link}
{kind=link}
Therapeutic targeting of macrophage dysfunction in asthma. A number of commonly prescribed therapies may beneficially modulate macrophage function in asthma. LTB4: leukotriene B4; IL: interleukin.
The recent paradigm shift in our understanding of the ontogeny and activation of various macrophage populations within the lung opens up new areas of basic and clinical asthma research. Open questions include: What are the asthma-specific alterations of resident, induced and infiltrating macrophage/monocyte populations (studies to date have focused on broad populations of alveolar/airway macrophages and/or peripheral monocytes)? Do early life events associated with predisposition to subsequent development of asthma alter the macrophage development and landscape of the lungs and airways irrevocably and do these changes drive pathology? Can these various subtypes of macrophage be specifically targeted for therapeutic gain? What is the role of infiltrating monocytes in asthmatic airways inflammation in both stable and exacerbating phenotypes and what is the fate of macrophage/monocyte populations during periods of inflammatory resolution?
At present, a lack of bona fide markers for the distinction and isolation of these newly recognised various macrophage subsets is the biggest hindrance to the progress of research in this field; however, continued combination of macrophage fate mapping with “omics” approaches in the mouse may be expected to provide the tools to answer these questions. In the future, accurate macrophage phenotyping may play a role in the development of targeted personalised treatment programmes and could guide selection of optimal asthma therapies in the clinic.
Disclosures
Acknowledgements
The authors would like to thank Steven Maltby (University of Newcastle, New Lambton Heights, NSW, Australia) for providing valuable feedback during manuscript preparation.
Footnotes
Support statement: Michael Fricker is supported by the National Health and Medical Research Council (NHMRC) Centre of Excellence for Severe Asthma. Peter G. Gibson is supported by a NHMRC practitioner fellowship. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received January 27, 2017.
- Accepted June 11, 2017.
- Copyright ©ERS 2017
References










