





Abstract
A proportion of pulmonary arterial hypertension (PAH) patients do not reach treatment goals with phosphodiesterase-5 inhibitors (PDE5i). RESPITE investigated the safety, feasibility and benefit of switching from PDE5i to riociguat in these patients.
RESPITE was a 24-week, open-label, multicentre, uncontrolled study. Patients in World Health Organization (WHO) functional class (FC) III, with 6-min walking distance (6MWD) 165–440 m, cardiac index <3.0 L·min−1·m−2 and pulmonary vascular resistance >400 dyn·s·cm−5 underwent a 1–3 day PDE5i treatment-free period before receiving riociguat adjusted up to 2.5 mg maximum t.i.d. Exploratory end-points included change in 6MWD, WHO FC, N-terminal prohormone of brain natriuretic peptide (NT-proBNP) and safety.
Of 61 patients enrolled, 51 (84%) completed RESPITE. 50 (82%) were receiving concomitant endothelin receptor antagonists. At week 24, mean±sd 6MWD had increased by 31±63 m, NT-proBNP decreased by 347±1235 pg·mL−1 and WHO FC improved in 28 patients (54%). 32 patients (52%) experienced study drug-related adverse events and 10 (16%) experienced serious adverse events (2 (3%) study drug-related, none during the PDE5i treatment-free period). Six patients (10%) experienced clinical worsening, including death in two (not study drug-related).
In conclusion, selected patients with PAH may benefit from switching from PDE5i to riociguat, but this strategy needs to be further studied.
Abstract
Switching to riociguat in PAH patients with inadequate response to PDE5i improved exercise capacity and NT-proBNP http://ow.ly/e6xL30dXCgy
Introduction
Pulmonary arterial hypertension (PAH) is characterised by an angioproliferative pulmonary vasculopathy that mainly affects the precapillary arterioles and results in progressive obliteration of the pulmonary vascular bed [1]. As a result, the workload on the right ventricle gradually increases, eventually resulting in right-sided heart failure and death if left untreated [2].
PAH remains an incurable disease, but several treatments have been developed that improve symptoms and outcomes [3, 4]. The current treatment objective for patients with PAH is to achieve a low-risk status according to the 2015 European Society of Cardiology (ESC)/European Respiratory Society (ERS) treatment guidelines [3]. PAH treatment can be initiated as monotherapy or as combination therapy with drugs targeting different pathways [4].
Contemporary treatments for PAH target three pathways: the prostacyclin pathway, the endothelin pathway and the nitric oxide (NO)-soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP) pathway [3, 5]. Three drugs targeting the NO-sGC-cGMP pathway are approved for the treatment of PAH: two phosphodiesterase-5 inhibitors (PDE5i), sildenafil and tadalafil; and the sGC stimulator, riociguat [6–8]. From a global perspective, PDE5i are the most frequently used treatments for PAH, either as monotherapies or combination regimens, particularly with endothelin receptor antagonists (ERAs) [9–14]. Riociguat and PDE5i target different molecular targets in the same pathway, and concomitant administration of riociguat with a PDE5i is contraindicated. This is mainly based on the open-label extension of the PATENT-PLUS study, which revealed an increased risk of side-effects, most importantly systemic hypotension, over the long term, and no evidence of a positive benefit/risk ratio [15–18].
Clinical data indicate that a sizeable proportion of patients receiving PDE5i do not reach treatment goals. For example, in the AMBITION study, 73% of patients with PAH receiving tadalafil monotherapy and 61% of those receiving tadalafil in combination with ambrisentan did not achieve a satisfactory clinical response at week 24 of the study [19]. Furthermore, in the SERAPHIN study, event-free survival of patients receiving PDE5i monotherapy was approximately 50% at 3 years [20].
For patients without a sustained response to PDE5i, there is a biological rationale for switching to sGC stimulators. In healthy subjects, cGMP production in pulmonary vascular smooth muscle cells (PVSMCs) arises from activation of sGC by NO released from neighbouring endothelial cells [21]. Phosphodiesterase-5 (PDE5) mediates degradation of cGMP in the pulmonary circulation. Inhibiting PDE5 activity increases intracellular cGMP levels in situations of impaired NO signalling, resulting in vasodilation and inhibition of PVSMC proliferation [22]. However, in patients with PAH, evidence suggests that NO bioavailability is reduced. During disease progression, there is a decline in endothelial function, resulting in NO depletion and low intracellular cGMP concentrations, which may render PDE5i ineffective [21, 23–26]. Riociguat directly stimulates sGC, increasing intracellular cGMP in the presence or absence of NO [27, 28]. Based on this mode of action, riociguat may benefit patients with an insufficient response to PDE5i.
The Riociguat clinical Effects Studied in Patients with Insufficient Treatment response to PDE5 inhibitors (RESPITE) study [29] was designed to investigate whether it is safe, feasible and beneficial to replace PDE5i with riociguat in patients with PAH who have an inadequate response to PDE5i.
Methods
Study design
RESPITE was a 24-week, prospective, exploratory, open-label, multicentre, uncontrolled, single-arm study including patients from nine countries in Europe and North America (ClinicalTrials.gov Identifier NCT02007629) [30].
Patients
Men and women aged 18–75 years with symptomatic PAH (including idiopathic PAH, heritable PAH, drug- and toxin-induced PAH, and PAH associated with congenital heart disease) and an insufficient response to stable treatment with tadalafil (40 mg once daily) or sildenafil (minimum 20 mg three times daily (t.i.d.), maximum 80 mg t.i.d.) for ≥90 days were included. Patients were permitted to continue background therapy with ERAs if they had been receiving stable treatement for ≥90 days. Final inclusion criteria were: World Health Organization functional class (WHO FC) III, 6-min walking distance (6MWD) 165–440 m, cardiac index <3.0 L·min−1·m−2, mean pulmonary artery pressure (mPAP) >30 mmHg, pulmonary artery wedge pressure ≤15 mmHg and pulmonary vascular resistance (PVR) >400 dyn·s·cm−5. The first 30 patients were recruited with narrower haemodynamic inclusion criteria (cardiac index <2.5 L·min−1·m−2 and PVR >480 dyn·s·cm−5), which were later amended in order to enhance the feasibility of the trial. Exclusion criteria are described in the supplementary material.
The institutional review board at each participating centre approved the protocol. The study was carried out in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All patients provided written informed consent.
Study procedures
There was a 14-day screening period while patients were still receiving PDE5i, during which baseline measurements were obtained. PDE5i were stopped 1–3 days before the first dose of riociguat to allow a PDE5i treatment-free period of 24 h for patients receiving sildenafil and 72 h for patients receiving tadalafil (figure S1) [15, 16]. Patients receiving ERAs continued with their regular dose, and other supportive measures were also continued. Riociguat was started at a daily dose of 1.0 mg t.i.d. During an 8-week dose-adjustment phase, riociguat doses were individually increased according to the approved riociguat dose-adjustment scheme, i.e. in 0.5 mg steps every 2 weeks until the highest tolerated dose, or up to a maximum dose of 2.5 mg t.i.d. [15, 16]. The individualised optimal dose of riociguat reached at the end of this phase was continued for another 16 weeks. Patients were seen at weeks 0, 2, 4, 6 and 8 during the dose-adjustment phase and at weeks 12 and 24 during the maintenance phase.
Patients underwent right heart catheterisation at baseline (while still receiving PDE5i) and again at week 24. 6MWD, WHO FC and N-terminal prohormone of brain natriuretic peptide (NT-proBNP) concentration were assessed at every visit. Measurements taken during screening were used as baseline, which was the last documented value while still receiving PDE5i. Week 0 was the last documented value before starting riociguat (after the PDE5i treatment-free period).
At the end of the study, patients could participate in an extended drug-supply phase for 18 months or until reimbursement. If a patient discontinued study drug prematurely or did not enter the extended drug-supply phase of the study, there was a 30-day safety follow-up phase.
Outcome measures
All end-points were exploratory and included: change from baseline to week 24 in 6MWD, NT-proBNP, WHO FC, pulmonary haemodynamics and EuroQol 5-Dimensions questionnaire (EQ-5D) score (quality of life); the proportion of patients experiencing clinical worsening; and safety and tolerability. A combined responder end-point was also assessed. Responders were defined as patients who at week 24 were free from clinical worsening, achieved WHO FC I/II and had a ≥30 m increase in 6MWD.
Clinical worsening was defined as: death; atrial septostomy; lung transplantation; non-planned PAH-related hospitalisation; start of new PAH treatment (ERA, inhaled or oral prostanoid) or modification of pre-existing treatment, initiation of intravenous or subcutaneous prostanoids; persistent decrease of >15% from baseline or >30% from last measurement in 6MWD; persistent worsening of WHO FC; or appearance or worsening of signs/symptoms of right heart failure not responding to optimised oral diuretic therapy. All identified and suspected clinical worsening events were confirmed by independent central adjudication. Details of the statistical analysis are given in the supplementary material.
Results
Patients
Of 79 patients screened, 61 patients were enrolled in RESPITE (figure 1). Most patients were diagnosed with idiopathic PAH (n=56, 92%) and all were in WHO FC III (table 1). Before entering RESPITE, 40 patients (66%) were receiving sildenafil and 21 (34%) were receiving tadalafil. 14 patients (23%) had been receiving PDE5i treatment for ≤6 months before entering RESPITE, and 47 patients (77%) had been receiving PDE5i treatment for >6 months (maximum: 10 years). At baseline, 50 patients (82%) were receiving combination therapy with ERAs (23 (38%) ambrisentan, 16 (26%) bosentan and 11 (18%) macitentan). 45 patients (74%) were receiving concomitant diuretic treatment at baseline and two patients (3%) commenced loop diuretics during the study.

Patient disposition and flow in RESPITE over 24 weeks of treatment. #: primary reason for discontinuation; ¶: two patients died during the main study (days 27 and 49). Of the two deaths during the study period, one was attributed to pneumonia (after withdrawal by patient, the primary reason for discontinuation) and one to subdural haematoma.
Patient characteristics at RESPITE baseline (the last documented value while still receiving phosphodiesterase-5 inhibitor)
Of the 61 patients enrolled in RESPITE, 51 (84%) completed 24 weeks of treatment and 10 (16%) discontinued treatment. Primary reasons for discontinuation included four adverse events, one death, one lack of efficacy, one physician withdrawal and three withdrawals by patient (one of whom subsequently died; see below). At week 24, of 51 patients remaining in the study, 47 (92%) were receiving the maximum dose of riociguat (2.5 mg t.i.d.). The remaining four patients (8%) were receiving riociguat 2.0 mg t.i.d.
Efficacy
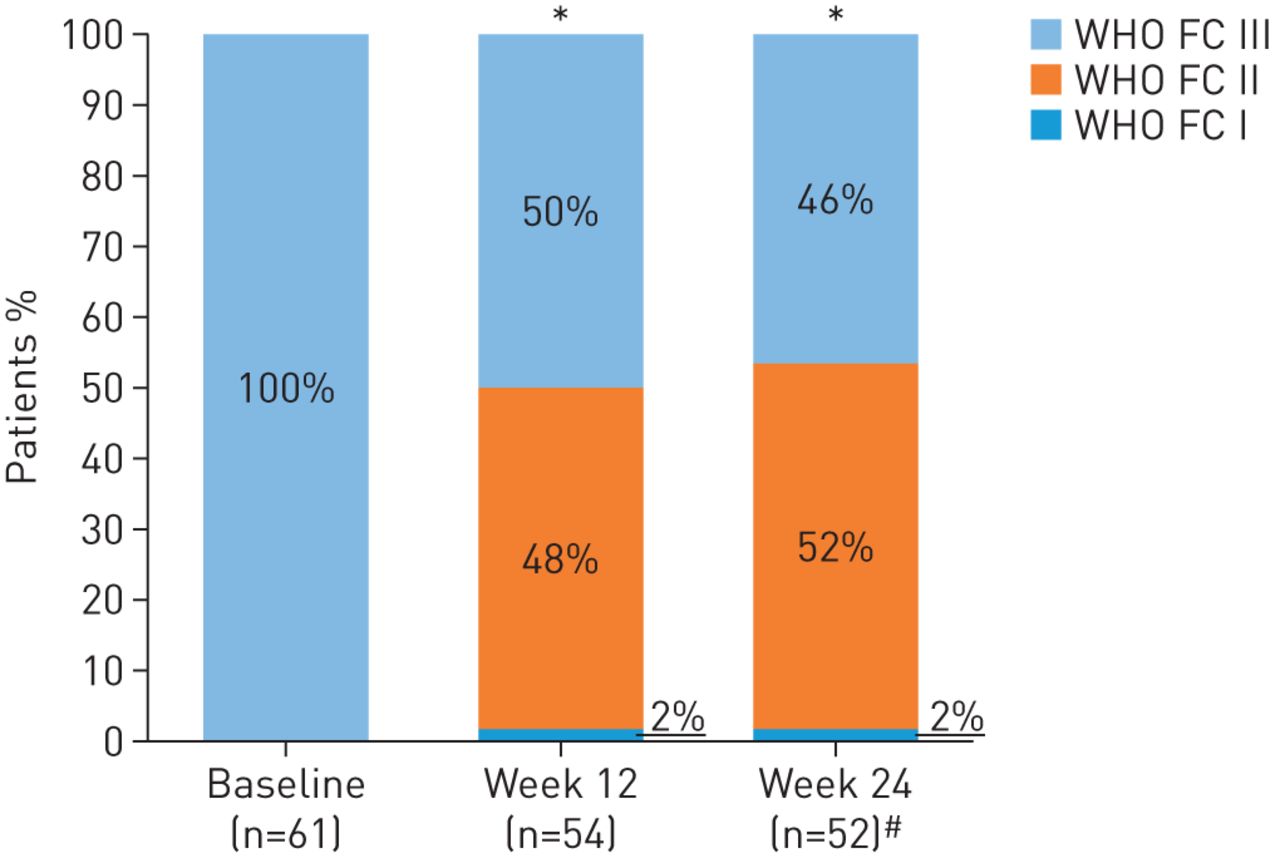
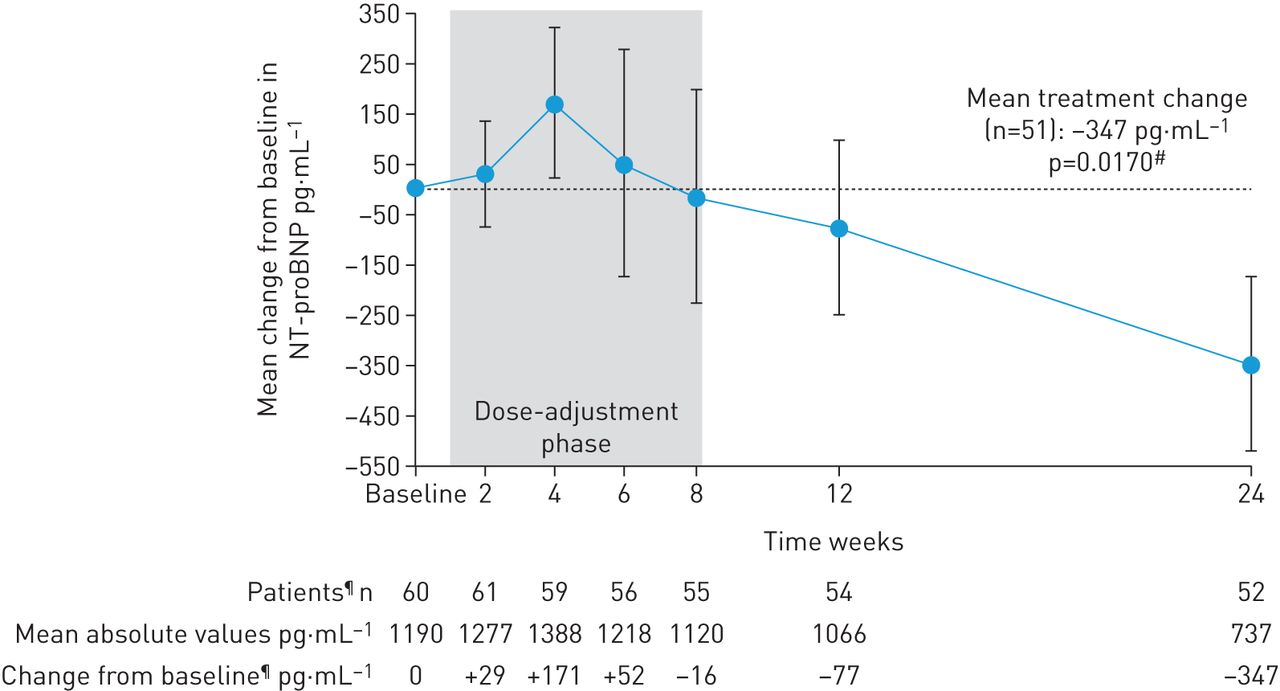
In patients completing 24 weeks’ treatment, mean±sd 6MWD increased from baseline by +31±63 m (95% CI 13–49 m; p=0.0010; figure 2). Mean±sd NT-proBNP levels decreased by –347±1235 pg·mL−1, a relative reduction of 22% (95% CI 5−37%; p=0.0170; figure 3). At week 24, 28/52 patients (54%) had improved WHO FC (52% to WHO FC II, 2% to WHO FC I; p<0.0001; figure 4). Haemodynamic variables for the entire population at baseline are shown in table 1. Pairwise values at baseline and week 24 for haemodynamic variables were available for 49 patients. In these patients, at week 24, mean±sd PVR was reduced by –103±296 dyn·s·cm–5 (95% CI –188 to –18 dyn·s·cm–5; p=0.0184), mean±sd mPAP was reduced by –2.8±8.8 mmHg and right atrial pressure was reduced by –0.8±4.2 mmHg. Mean±sd cardiac index and mixed venous oxygen saturation increased by +0.3±0.5 L·min−1·m−2 (95% CI 0.2–0.5 L·min−1·m−2; p=0.0001) and +1.0±6.3%, respectively (table 2). An additional sensitivity analysis was performed to address the potential bias arising from the 10 patients who did not complete the study. A worst possible value was imputed for patients who died or withdrew with no termination visit for 6MWD, WHO FC, and EQ-5D, the last observation was carried forward (LOCF) for pulmonary haemodynamics and the last post-baseline measurement was imputed for NT-proBNP. This analysis still showed a change from baseline for all parameters in agreement with the observed study results in the patients who completed the study. Although the change from baseline was lower and not statistically significant for 6MWD and NT-proBNP, it remained significant for change in WHO FC (table 2; tables S1 and S2).
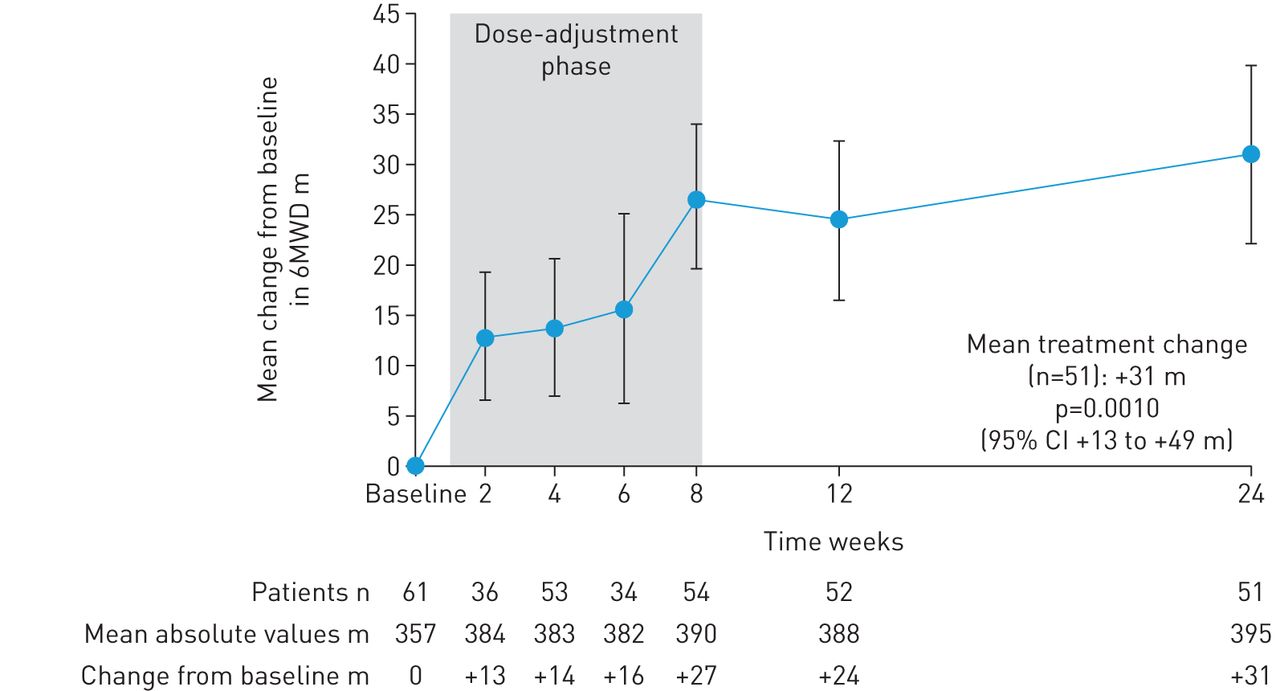
Change from baseline (the last documented value while still receiving phosphodiesterase-5 inhibitor) in 6-min walking distance (6MWD) over time in RESPITE. Data are mean±sem; observed values.
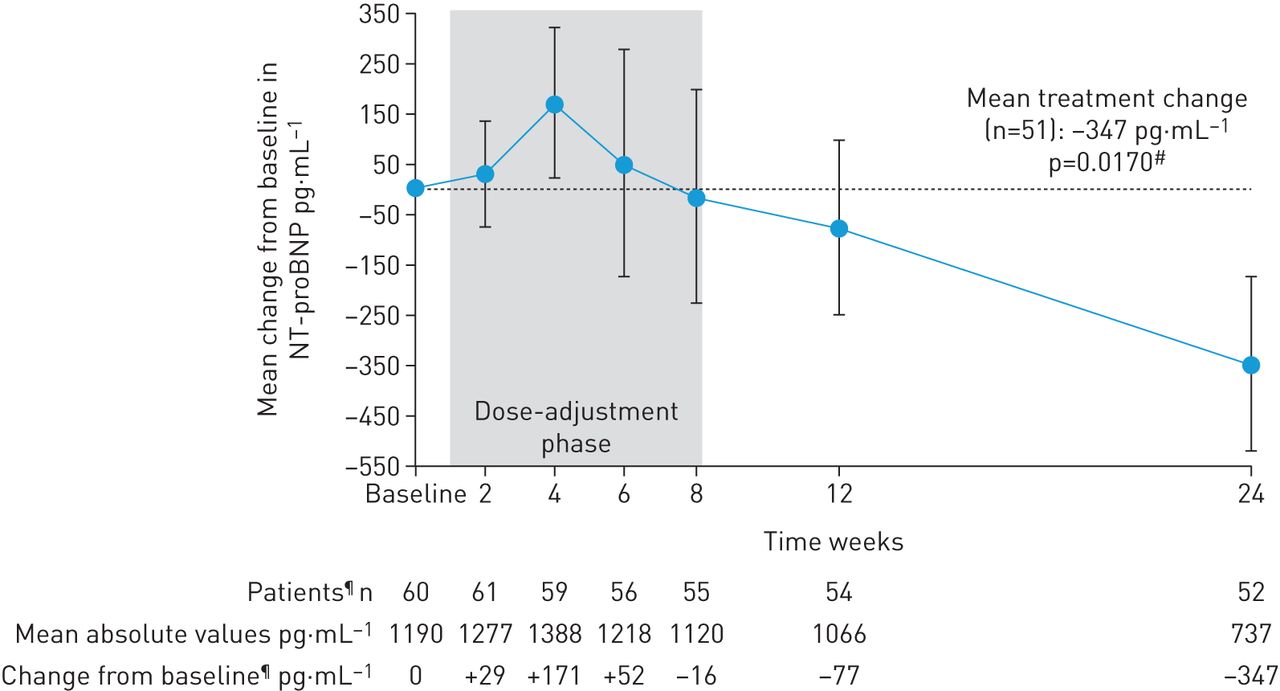
Change from baseline (the last documented value while still receiving phosphodiesterase-5 inhibitor) in N-terminal prohormone of brain natriuretic peptide (NT-proBNP) over time in RESPITE. Data are mean±sem; observed values. #: p-value calculated for relative change from baseline; ¶: value missing for one patient at baseline; n=52 for change from baseline at week 24 due to one patient withdrawal at day +159 having undergone a week 24 assessment of NT-proBNP.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
World Health Organization functional class (WHO FC) at baseline (the last documented value while still receiving phosphodiesterase-5 inhibitor), week 12 and week 24 in RESPITE. Data are observed values. *: p<0.0001, change from baseline to weeks 12 and 24; #: n=52 due to one patient withdrawal at day +159 having undergone a week 24 assessment of WHO FC.
Change in haemodynamics from baseline (the last documented value while still receiving phosphodiesterase-5 inhibitor) to week 24 and last-observation-carried-forward (LOCF) sensitivity analysis
At week 24, 16/51 patients (31%) met the combined responder end-point of freedom from clinical worsening, WHO FC I/II and an improvement in 6MWD ≥30 m. A sensitivity analysis that included patients who completed or withdrew from the study (n=60) showed that 16 patients (27%) achieved the combined responder end-point. Change from baseline to week 24 in EQ-5D scores, and worst-case imputed values for EQ-5D are presented in table S2. A post hoc analysis was performed to determine whether patients had changed their ESC/ERS risk category at week 24, with an overall low-risk profile assumed when >50% of the available variables met the low-risk thresholds. This was found to be the case in 25 of 49 patients with determinable status (51%) at week 24 compared with 9 patients (15%) at baseline. However, it should also be noted that 3 patients (6%) had a high-risk profile at week 24. A sensitivity analysis taking into account that 10 patients had discontinued prematurely from the study resulted in a rate of 41% of patients from the overall population who achieved a low risk status at week 24. The effects of riociguat on 6MWD, NT-proBNP and haemodynamics appeared to be independent of the previous type of PDE5i treatment (figure S2) or concomitant ERA use (tables S3 and S4; figure S3).
Clinical worsening
Six patients (10%) experienced one or more defined and adjudicated events of clinical worsening: two deaths (see below), two patients who started a new PAH treatment, two patients who experienced persistent worsening of 6MWD due to PAH and one patient who experienced signs/symptoms of right heart failure that did not respond to optimised oral diuretic therapy. No clinical worsening events occurred during the PDE5i treatment-free period.
Safety
Adverse events
During the study, 58 patients (95%) experienced an adverse event, the most frequent of which are described in table 3. Four patients (7%) experienced adverse events leading to discontinuation of study drug, including two patients (3%) with right ventricular failure (days +15 and +158 after starting riociguat treatment), one patient (2%) with asthenia (day +2) and one patient (2%) with symptomatic hypotension (day +16). Of the two patients experiencing right ventricular failure, one concurrently experienced renal failure and asymptomatic hypotension, and the other concurrently experienced dyspnoea. Patients who did not enter the extended drug-supply phase of the study or discontinued the study prematurely underwent a 30-day safety follow-up. 12 patients (20%) experienced adverse events during the 30-day safety follow-up, and one patient (2%) experienced a serious adverse event of cholecystitis. The most common adverse events during the follow-up period were nasopharyngitis (n=3; 5%) and peripheral oedema (n=2; 3%).
Most frequently reported adverse events, adverse events of special interest and serious adverse events
Adverse events of special interest
Syncope was not reported during RESPITE. 10 patients (16%) experienced adverse events of hypotension, seven of whom experienced symptomatic hypotension; six events were considered mild, three moderate and one severe. Hypotension led to dose reduction or interruption in two patients and drug discontinuation in two patients. SBP <95 mmHg was measured in three patients at week 0, four at week 6, three at week 12 and five at week 24. Mean systolic and diastolic blood pressures and heart rate during the study are shown in figure S4.
There was one event of haemoptysis, which was considered mild, not study drug-related and was described as cough with occasional blood spots. Therapeutic intervention was not required and the patient continued study medication without experiencing additional episodes of haemoptysis.
Serious adverse events
Serious adverse events occurred in 10 patients (16%). There were three cases of right ventricular failure, and one case each of lower abdominal pain, dyspepsia, asthenia, cholecystitis, pneumonia, subdural haematoma, depression, renal failure, interventional procedure and hypotension (moderate in severity). Two serious adverse events were considered by the investigators to be study drug-related: right ventricular failure and asthenia. No serious adverse events were reported during the PDE5i treatment-free period, and only two patients (3%) reported serious adverse events during the first 2 weeks of riociguat treatment.
Deaths
Two patients (3%) died during the study: one death was attributed to pneumonia on day +55 after the patient had discontinued riociguat treatment on day +28 (reason for discontinuation: patient withdrawal); the other death was attributed to subdural haematoma following a fall (day +50, not related to hypotension). Prior PDE5i therapy was sildenafil for the patient who died of pneumonia and tadalafil for the patient who experienced a subdural haematoma. No deaths were considered study drug-related.
Discussion
RESPITE was the first study to investigate potential clinical improvement by switching therapies that target different molecules in the same signalling pathway in PAH and has provided preliminary evidence that this approach may be not only tolerable and safe, but also potentially beneficial in PAH patients with an insufficient response to their previous treatment, which consisted of PDE5i monotherapy or PDE5i/ERA combination therapy. In total, 51 of 61 patients (84%) completed the 24-week study, and in these patients, replacing PDE5i with riociguat resulted in improvements in haemodynamic variables, cardiac biomarkers and clinical end-points including 6MWD and WHO FC. The improvements in 6MWD and EQ-5D were comparable to the minimally important differences for clinical relevance previously reported for these parameters [29, 31, 32]. However, 10 patients (16%) did not complete the study, mostly due to side-effects or clinical worsening. Overall, 10 patients (16%) experienced serious adverse events; 10 patients (16%) had episodes of hypotension (seven symptomatic); and six patients (10%) experienced clinical worsening events. No serious adverse events or clinical worsening events occurred during the PDE5i treatment-free period, and no new safety signals were identified.
The drop-out rate in RESPITE was higher than that of the 2.5 mg maximum arm in the pivotal PATENT-1 study (16% versus 7% for RESPITE and PATENT-1, respectively). However, the baseline characteristics were also substantially different, with 100% of patients in WHO FC III in RESPITE versus 55% in the PATENT-1 2.5 mg maximum arm, and 48% of patients in the PATENT-1 2.5 mg maximum arm being treatment-naïve. Therefore, direct comparison between these groups may not be informative.
The majority of enrolled patients (82%) had been receiving combination therapy with PDE5i and ERAs before inclusion, and 74% of the population were receiving diuretics at baseline. Despite stable and prolonged pretreatment, all patients had severe haemodynamic impairment, were in WHO FC III and had a 6MWD <440 m. According to the 2015 European pulmonary hypertension treatment guidelines, these patients would have been classified as intermediate risk, which is considered an inadequate response to therapy [3]. It should be noted that at week 24 of RESPITE, 41% of the overall population (25/61) would have been considered to have an overall low risk profile (where >50% of variables were low risk), compared with 15% at baseline.
The potential direct interaction between bosentan and sildenafil that results in reduced plasma levels of the latter [33] has been suggested as a possible explanation for why the COMPASS-2 study, which added bosentan to sildenafil, did not achieve its primary end-point [34]. However, in RESPITE there was no indication that type of ERA or prior PDE5i therapy affected 6MWD, NT-proBNP, cardiac index or PVR.
Although not mechanistically studied, the findings of RESPITE support the hypothesis that a defective NO-sGC-cGMP pathway might explain why some patients have no sufficient or sustained response to PDE5i therapy. In such patients, direct stimulation of sGC may be more effective than inhibition of PDE5, but this hypothesis is still unproven. The continued improvements seen in 6MWD from baseline up to week 24 of RESPITE may support this theory. NT-proBNP levels decreased substantially over the study period, although interestingly, levels temporarily increased in the period between the end of PDE5i therapy and the individually optimised dose of riociguat. This observation suggests that the PDE5i were still having a positive effect in the study population, albeit not enough for patients to reach or maintain treatment goals. A PDE5i treatment-free period of 24 h for sildenafil and 72 h for tadalafil was used before the start of riociguat treatment to avoid an exposure overlap, and riociguat was adjusted over an 8-week period. Although no clinical worsening events occurred during the PDE5i treatment-free period, stopping treatment remains a matter of concern, especially in patients receiving PDE5i monotherapy. Shorter switching periods might be preferable and future studies need to address this question as well as whether starting riociguat at higher doses with shorter dose-adjustment periods is safe and feasible. Furthermore, the three times daily dosing interval with riociguat compared with once daily dosing of tadalafil may affect patient adherence to treatment.
It is also unclear whether patients who are likely to have a more favourable response to sGC stimulators than to PDE5i can be identified a priori. Biomarkers of NO synthesis have been shown to correlate with the severity of PAH [24, 35] and may be used to identify patients with impaired NO signalling who could benefit from sGC stimulation.
Given the exploratory and uncontrolled nature of RESPITE, various end-points were pre-specified but no primary end-point was defined. The study intended to provide preliminary signals of safety and efficacy before embarking on a larger, randomised, controlled trial, as agreed a priori between the sponsor and steering committee. As there were no new safety signals, and improvements from baseline in 6MWD, WHO FC, NT-proBNP levels and haemodynamics, a randomised, controlled trial to further investigate the strategy of switching from PDE5i to riociguat is under way (Riociguat rEplacing PDE-5i Therapy evaLuated Against Continued PDE-5i thErapy (REPLACE); clinicaltrials.gov identifier NCT02891850).
Strengths of the present study include the prospective, multicentre study design; a relatively homogenous patient population; the use of systematic safety and efficacy assessments, including invasive haemodynamics at entry and at the end of the study; and the central, independent adjudication of clinical worsening events. The most important limitations of RESPITE are the absence of a control group, which precludes speculation on whether switching affects long-term survival, and the lack of blinding. Additional limitations include the small sample size, and the relatively high drop-out rate (16%), which may have introduced a bias to the analysis. We have attempted to address this potential bias with a sensitivity analysis using worst possible values for 6MWD, WHO FC and EQ-5D, as well as LOCF for pulmonary haemodynamics, in order to provide the most conservative view of the data. Improvement was still in line with the overall results of the study, but no longer statistically significant for 6MWD and NT-proBNP, with the change for WHO FC remaining significant. Other limitations include the lack of a long-term continuation phase, and the absence of mechanistic data allowing identification of patients likely to respond or not respond to switching. Two deaths were observed in this study, which might raise concerns, although neither of the deaths (one due to pneumonia and one due to subdural haematoma) was considered by the investigators to be study drug-related or due to worsening PAH. Given the lack of a control group and the rate of study withdrawals and clinical worsening events, further evaluation to clarify the safety of switching is required. We also acknowledge that the patients enrolled in this study could have been treated with double or triple sequential combination therapy as recommended in current guidelines and that comparisons with these treatment strategies cannot be made. Finally, RESPITE predominantly included patients with idiopathic PAH (92%) so the findings cannot be extrapolated to other forms of PAH.
Conclusions
The findings from RESPITE indicate that replacing PDE5i with riociguat may be a feasible and effective treatment strategy for patients with PAH who have an insufficient clinical response to PDE5i monotherapy or to combination therapy with ERA and PDE5i. This study represents an important step towards determining if this new treatment strategy is an effective approach to the management of patients with PAH, although additional data from larger, randomised, controlled studies are needed to further establish the safety and efficacy of this approach.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02425-2016_Supplement
Disclosures
Supplementary Material
R.L. Benza ERJ-02425-2016_Benza
D. Busse ERJ-02425-2016_Busse
M. Chang ERJ-02425-2016_Chang
P. Colorado ERJ-02425-2016_Colorado
P.A. Corris ERJ-02425-2016_Corris
H-A. Ghofrani ERJ-02425-2016_Ghofrani
E. Grünig ERJ-02425-2016_Grunig
M.M. Hoeper ERJ-02425-2016_Hoeper
P. Jansa ERJ-02425-2016_Jansa
J.R. Klinger ERJ-02425-2016_Klinger
D. Langleben ERJ-02425-2016_Langleben
C. Meier ERJ-02425-2016_Meier
R. Naeije ERJ-02425-2016_Naeije
S. Rosenkranz ERJ-02425-2016_Rosenkranz
G. Simonneau ERJ-02425-2016_Simonneau
C.D. Vizza ERJ-02425-2016_Vizza
Acknowledgements
Editorial support was provided by Adelphi Communications Ltd, Bollington, UK, supported by Bayer AG. The authors wish to thank the members of the RESPITE Clinical Event Committee, who served as independent adjudicators of clinical worsening events.
Footnotes
This article has supplementary material available from erj.ersjournals.com
This study is registered at ClinicalTrials.gov with identifier NCT02007629.
Support statement: This study was supported by Bayer AG, Berlin, Germany. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received December 9, 2016.
- Accepted July 12, 2017.
- Copyright ©ERS 2017
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










