





Abstract
Lymphoedema, myelodysplasia, fibrosis and pulmonary alveolar proteinosis complicating a novel GATA2 mutation http://ow.ly/RHci303h8ks
To the Editor:
Pulmonary alveolar proteinosis (PAP) is a rare syndrome characterised by an abnormal accumulation of surfactant proteins in pulmonary alveoli which impairs gas exchange [1]. PAP is classified into three groups: autoimmune PAP (aPAP), defined by the presence of anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) autoantibodies, secondary PAP (sPAP) and genetic PAP. Haematological diseases are the most common causes of sPAP, particularly myelodysplastic syndrome (MDS) [1, 2]. The diagnosis of PAP is based on a chest computed tomography (CT) scan, which demonstrates a crazy-paving pattern, and a typical positive periodic acid–Schiff (PAS) staining of bronchoalveolar lavage (BAL) [3]. Nowadays, a surgical lung biopsy is seldom needed to diagnose PAP, except for atypical presentation as in the case described below with concomitant pulmonary fibrosis and sPAP in a patient with GATA2 (GATA binding protein 2) deficiency.
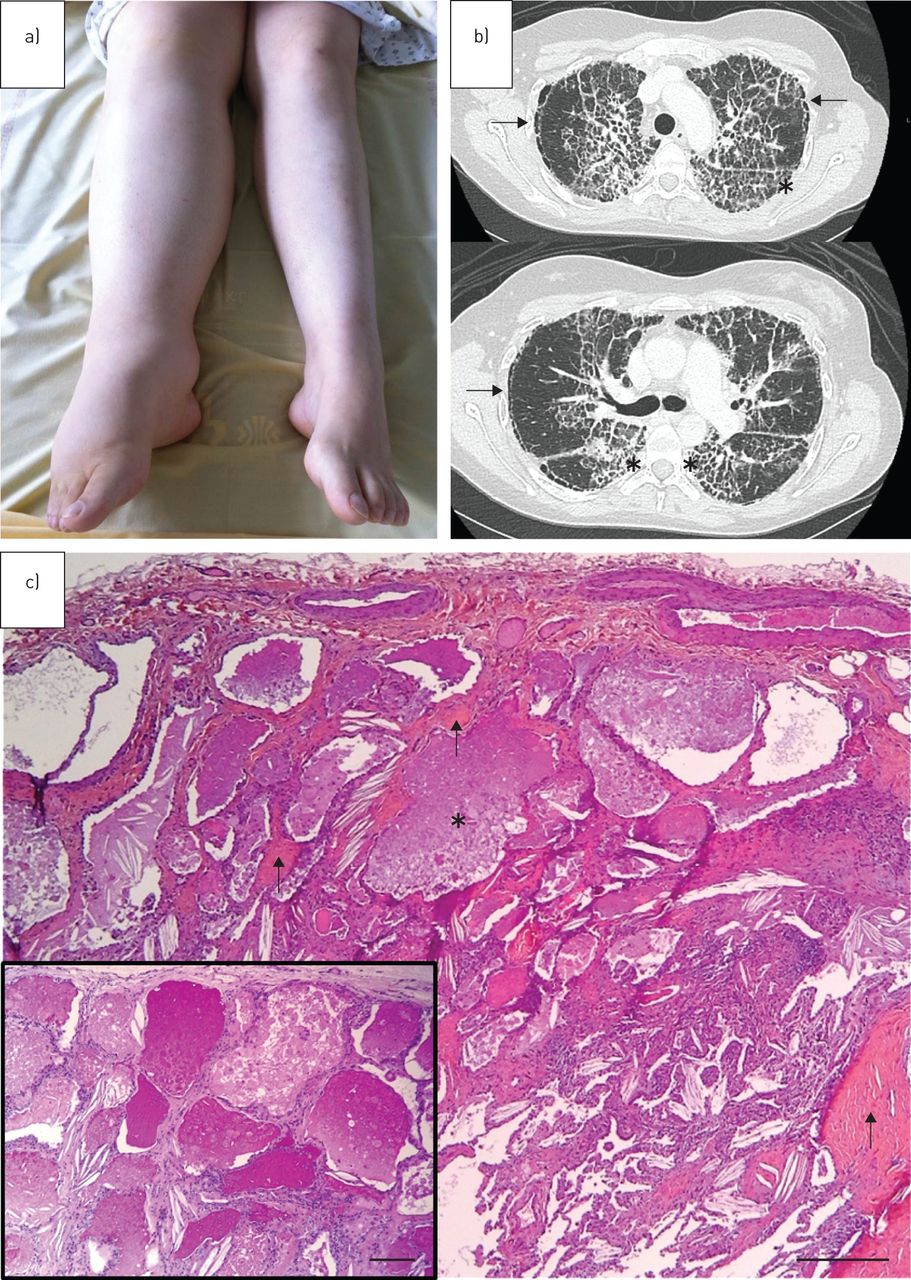
A 28-year-old Caucasian woman from North Africa, a nonsmoker, an executive in a fuel company, was admitted to the Respiratory Dept of Pontchaillou Hospital (Rennes, France) in November 2013 with increasing New York Heart Association (NYHA) stage 2 dyspnoea, dry cough and interstitial lung disease. She had no significant family history and no past medical history except for right lower limb idiopathic lymphoedema diagnosed 5 years earlier (figure 1a). Digital clubbing was found on clinical examination as well as a normal chest auscultation. Retractile reticular opacities in both lung apices were noted on chest radiography. A chest CT scan showed diffuse bilateral thickening of the interlobular and intralobular septae predominant in the upper lobes, associated with profuse subpleural reticulations (figure 1b). Spirometry demonstrated a pure restrictive pattern: total lung capacity 68% of predicted and forced vital capacity (FVC) 52% of predicted. The transfer factor of the lung for carbon monoxide (TLCO) was reduced (40% of predicted). BAL was opalescent and found to contain 50 000 cells·mL−1 with 72% macrophages and 26% lymphocytes. PAS staining could not be performed on the BAL and a surgical lung biopsy was completed. Histological findings demonstrated a markedly thickened visceral pleura, and prominent subpleural and paraseptal fibrosis. Interstitial fibrosis accompanied alveoli filled with an eosinophilic, proteinaceous material which proved to be PAS-positive and diastase-resistant. Vacuolated, foamy alveolar macrophages and cholesterol crystals were also common. Rare fibroblastic foci and mild chronic inflammation were noted. Honeycombing, lymphoid aggregates, bronchiolisation, smooth muscle hyperplasia, squamous metaplasia, fibroelastosis and granulomas were absent (figure 1c). No anti-GM-CSF antibodies were identified in the sera. Haematological sPAP was suspected due to cytopenias: haemoglobin 10 g·dL−1, platelets 50 g·L−1, leukocytes 3.9 g·L−1, neutrophils 3.34 g·L−1, eosinophils 0.04 g·L−1, basophils 0.15 g·L−1, lymphocytes 0.33 g·L−1 and monocytes 0.04 g·L−1. Lymphocyte phenotype showed global B, T and natural killer lymphopenia. Gammaglobulins were normal: 12.4 g·L−1. A MDS was confirmed with a bone marrow biopsy (multilineage dysplasia and <5% blasts with normal karyotype). Due to the association of the young age of the patient, lymphoedema, MDS and sPAP, GATA2 deficiency was looked for and was positive: exon 5 C.1020_1029dup: p.R344GfsX43. Over a 9-month period, the MDS remained stable although her dyspnoea symptoms deteriorated markedly, reaching NYHA stage 3 dyspnoea with a decrease in her pulmonary function tests (FVC dropping from 52% to 40% and TLCO dropping from 40% to 25% of the predicted values). Her chest radiograph, however, remained unchanged. A bilateral whole-lung lavage was performed in September 2014 without any effect on her respiratory symptoms, spirometry or chest CT scan. This led us to consider haematopoietic stem cell transplantation (HSCT). Her brother was HLA-matched and GATA2 deficiency was negative.
{kind=link}
a) Right limb lymphoedema. b) Chest computed tomography image showing diffuse, bilateral and irregular thickening of the interlobular and intralobular septae clearly predominant in the upper lobes and apical segments of the lower lobes (asterisks). In the subpleural region of the upper lobes, we observed paraseptal emphysema (arrows). c) Surgical lung biopsy. Note the eosinophilic, granular, proteinaceous material within alveolar spaces characteristic of pulmonary alveolar proteinosis (asterisk). This material is periodic acid–Schiff (PAS)-positive (inset). Associated interstitial fibrosis is shown by arrows. Haematoxylin–eosin–safran: scale bar=500 µm; PAS (inset): scale bar=250 µm.
To the best of our knowledge, this is a novel frameshift GATA2 mutation and one of the rare descriptions of concomitant association of severe interstitial lung fibrosis with PAP in GATA2 deficiency at diagnosis [4]. It may be the end result of progressive chronic PAP or may be specific to PAP in patients with GATA2 deficiency. Marchand-Adam et al. [5] described a sequential case of a 35-year-old man with PAP secondary to idiopathic bone marrow aplasia, followed 10 years later by idiopathic pulmonary fibrosis associated with heterozygous deleterious germline TERT (telomerase reverse transcriptase) mutation. Similarly, aPAP was diagnosed in a 63-year-old woman that evolved into pulmonary fibrosis 7 years later [6]. The crazy-paving pattern was replaced by diffuse fibrotic changes, with a subpleural honeycombing pattern. The findings may suggest a surfactant dysfunction in the pathogenesis of fibrotic lung disease [6]. In the literature, the diagnosis, management and prognosis of sPAP differ from other types of PAP. The presentation on chest CT scan is more atypical and the prognosis of sPAP is poorer than aPAP (2-year survival of 46% versus 100%) [2, 3, 7, 8]. The management of sPAP is often based on the treatment of the underlying disease [1, 2]. Chemotherapy and early HSCT can cure sPAP. Whole-lung lavage is also an effective treatment to improve patient symptoms [1, 2, 4].
In this case report, even at a Competence Centre for Rare Pulmonary Diseases, pulmonologists and radiologists did not consider the PAP diagnosis in the first instance. The Respiratory Dept at Pontchaillou Hospital is experienced in managing PAP, with more than 60 whole-lung lavages performed in patients with aPAP in the last 15 years [1, 9]. PAS staining could not be performed on BAL afterwards. Transbronchial biopsies and especially cryobiopsies may be reliable diagnostic tools in interstitial lung diseases. However, thrombocytopenia led us to perform surgical lung biopsy, taking one single risk with easier bleeding management, in view of this atypical chest CT scan presentation. Indeed, the thickening of the septae in the upper lobes matching with the crazy-paving pattern, associated with profuse subpleural reticulations leading us to fibrosis, were unexpected findings. Compared with aPAP, sPAP is more difficult to diagnose, not only due to the atypical chest CT scan findings, but also because of the absence of anti-GM-CSF antibodies and the possible underlying diseases. The presentation is more diffuse with less geographical pattern in sPAP than in aPAP: 62% versus 19% and 24% versus 71%, respectively [8]. The subpleural ground-glass opacities and crazy-paving appearance are significantly less frequent in sPAP [8]. The use of surgical lung biopsy is more frequent in sPAP than aPAP (41% versus 7%) [7]. Thus, with an unclear interstitial lung disease, a PAS stain would be an interesting test to perform on BAL to avoid surgical lung biopsy.
In the cohort of Ishii et al. [2] of 31 patients with sPAP associated with MDS, the mortality rate was mainly due to the progression of PAP. Infections and the progression of the MDS were the second and third causes of mortality, respectively [2]. Only 10 out of 31 patients with MDS were treated with whole-lung lavage and only three had a positive response to the therapy [2].
GATA2 belongs to a family of zinc finger transcription factors that are critical regulators of gene expression in haematopoietic cells and GATA2 deficiency is a recent discovery [10]. GATA2 has been shown to regulate alveolar macrophage phagocytosis. In GATA2 deficiency, an alveolar macrophage dysfunction is present rather than a quantitative deficit given the abundance of alveolar macrophages in BAL fluid from patients affected by PAP [10].
Spinner et al. [10] described the clinical features of 57 patients with proven mutations leading to GATA2 deficiency. They identified a broad phenotype encompassing immunodeficiency, haematologic disorder, PAP and lymphatic dysfunction, which match with our patient. 54% were female with a median (range) age at first visit of 30 (4–76) years and median (range) age at initial presentation of 20 years (5 months to 78 years). Of the 50 patients who underwent bone marrow biopsies, 42 patients (84%) met diagnostic criteria for MDS with most bone marrow biopsies demonstrating multilineage dysplasia and <5% blasts. Chronic lymphoedema was present in six patients (11%). Biopsy-proven PAP was identified in 10 adults (18%) [10]. In the Discussion section of Spinner et al. [10], the authors report that the high prevalence of diffusion and ventilatory defects in their cohort may reflect alveolar filling, emphysematous changes, fibrosis or bronchiectasis secondary to recurrent pulmonary infection. However, no specific description of pulmonary fibrosis associated concomitantly with PAP has been described. The overall survival rate was 96% by age 20 years, 77% by age 40 years and 45% by age 60 years [10]. 21 patients underwent HSCT for MDS, PAP or recurrent infections. All patients with PAP demonstrated a significant improvement in pulmonary function [10, 11]. Importantly, family members should be screened routinely for GATA2 mutations before donating their bone marrow. In this case report, HSCT is now considered. We are expecting a new pulmonary evaluation before allograft. The discussion is underway with the patient, her family, the pulmonologists and the current transplant team assessing the risk/benefit balance. Indeed, mortality risk increases with TLCO or forced expiratory volume in 1 s ≤65% of predicted, NYHA stage 4 dyspnoea or oxygen therapy [1]. Otherwise, sPAP physiopathology supports a recurrence after lung transplantation because of alveolar macrophages quantitatively and qualitatively unable to provide surfactant clearance [4, 8].
In summary, this is one of the rare descriptions of concomitant association of severe pulmonary fibrosis and PAP at the diagnosis complicating a novel GATA2 mutation. This very rare syndrome is important to diagnose because of the discussion of HSCT before worsening of the respiratory status.
Acknowledgements
The authors thank Marie de Tayrac (Service de génétique moléculaire et génomique, hôpital Pontchaillou and UMR 6290 Institut de génétique et développement, Université Rennes 1, Rennes, France) for her help with molecular genetic databases.
Footnotes
Conflict of interest: None declared.
- Received February 2, 2016.
- Accepted August 9, 2016.
- Copyright ©ERS 2016
References










