





Abstract
Patients with symptomatic advanced chronic obstructive pulmonary disease (COPD) who experience recurrent exacerbations are particularly at risk of poor outcomes and present a significant burden on healthcare systems. The relative merits of treating with different inhaled combination therapies e.g. inhaled corticosteroids (ICS)/long-acting β2-agonist (LABA), LABA/long-acting muscarinic antagonists (LAMA), ICS/LABA/LAMA, in this patient group are poorly understood, as is reflected in current guidelines. The InforMing the PAthway of COPD Treatment (IMPACT) study will evaluate the efficacy and safety of fluticasone furoate (FF)/umeclidinium (UMEC)/vilanterol (VI) versus FF/VI or UMEC/VI over a 52-week treatment period. The study has been designed with a focus on understanding the comparative merits of each treatment modality in different phenotypes/endotypes.
This is a phase III, randomised, double-blind, three-arm, parallel-group, global multicentre study comparing the rate of moderate and severe exacerbations between FF/UMEC/VI and FF/VI or UMEC/VI over a 52-week treatment period. The study aims to recruit 10 000 patients from approximately 1070 centres. Eligible patients are aged ≥40 years, with symptomatic advanced COPD (Global initiative for chronic Obstructive Lung Disease (GOLD) group D) and an exacerbation in the previous 12 months.
The first patients were recruited to the IMPACT study (ClinicalTrials.gov: NCT02164513) in June 2014 and the anticipated completion date is July 2017.
Abstract
Treatment choices between LABA/LAMA versus ICS/LABA versus ICS/LABA/LAMA in GOLD group D subjects with COPD http://ow.ly/bg7R300C2uo
Introduction
Chronic obstructive pulmonary disease (COPD) is known to be the fourth leading cause of death worldwide [1, 2]. The economic burden of COPD in the USA alone is approximately $53.7 billion per year, with COPD exacerbations and severe COPD accounting for up to 84% of these costs [3].
COPD is characterised by chronic airflow limitation, resulting in progressively worsening symptoms, decreased activity, poor quality of life (QoL), exacerbations and premature death. Inhaled pharmacological maintenance therapy of chronic stable disease is largely limited to three classes of drugs: the long- and ultra-long-acting anti-muscarinic and β2-agonist bronchodilators and inhaled corticosteroids (ICSs) [4]. The recent Global initiative for chronic Obstructive Lung Disease (GOLD) 2015 strategy document has suggested considering treatment options on a two-axis continuum based on exacerbation risk and symptomatic progression [5]. The suggested pharmacological treatment paradigm is one of incremental additions of therapy depending on disease severity and progression. There is currently a paucity of randomised, controlled trial data supporting the various pharmacological treatment options available [3, 6], with the exception of the addition of ICSs [7] and oral phosphodiesterase type 4 (PDE4) inhibitors [8] for those at risk of exacerbations. Furthermore, there is no suggestion that targeting therapy towards any particular disease endotype improves outcomes, largely based on the absence of robust clinical information to date. However, large observational studies such as ECLIPSE (Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints) [9] and COPDGene [10] (GlaxoSmithKline, London, UK) are providing mounting evidence to help distinguish varying COPD phenotypes and potential markers of outcome, depending upon the underlying COPD biology. It appears that not all acute exacerbations are manifestations of the same pathobiological processes, and there is compelling evidence to divide them into infective and eosinophilic; episodes manifesting both pathologies are relatively rare [11]. There are now data to suggest that eosinophilic events are more responsive to corticosteroids, whereas this treatment may be detrimental in the events of infective origin. In addition, compelling data from recent post hoc analyses suggest that the reduction in exacerbation frequency seen with ICS treatment is directly related to the blood eosinophil count; there is reduced evidence of efficacy in patients with a count below 0.15×109 L [12, 13]. While ICSs decrease the risk of exacerbation in patients with COPD, they have been recognised as increasing the risk of pneumonia in GOLD D patients [14]. The risk of pneumonia may be increased further in patients with a low blood eosinophil count [15, 16]. Whether blood eosinophil count can be used to stratify patients so as to minimise the risk of pneumonia and maximise benefit is an important area for further study.
It is estimated that almost 50% of patients who are administered a long-acting inhaled drug receive an “open triple” ICS/long-acting muscarinic antagonists (LAMA)/long-acting β2-agonist (LABA) delivered via multiple inhalers [17–18]. Significant evidence exists to support the benefits to both lung function and symptoms of combining two bronchodilators. However, data for the positive effect on exacerbation prevention are limited to two large studies: the Effect of QVA149 versus NVA237 and Tiotropium on COPD Exacerbations (SPARK) study, which was designed to examine this question [19], and the Understanding Potential Long-term Impacts on Function with Tiotropium (UPLIFT) study, in which only 60% of patients received a LABA prior to randomisation to tiotropium [17]. There have been no prospective, randomised, controlled studies completed to date examining the benefits of dual bronchodilation (LAMA/LABA combinations) compared with ICS/LABA on exacerbations within an exacerbation-prone population. However, multiple recent studies with umeclidinium (UMEC), a LAMA, in addition to various ICS/LABAs, and similar studies with tiotropium and other LAMAs, have consistently shown benefit to lung function over and above the dual combination alone, similar to the effect size observed when adding a LAMA to a LABA monotherapy [20–22]. Extensive observational studies have suggested that triple therapy is effective in preventing exacerbations compared with dual therapy [23], which would seem to suggest that a net positive effect on exacerbations will be seen with triple therapy. There are robust data supporting the value of adding ICS therapy to LABA in preventing exacerbations [24, 25], but there are no randomised studies addressing the key question of whether or not ICS added to dual bronchodilation is likely to offer additional protection. Importantly, no prospective studies have assessed the endotypes in which ICS add-on therapy would have the best therapeutic index.
Rationale
Since the recent widespread regulatory approval of fluticasone furoate (FF)/vilanterol (VI), UMEC/VI and UMEC monotherapy, it has proven possible to formulate all three agents within one device [26], raising the possibility of benefits for adherence levels and ease of switching between different classes and combinations (without changing the delivery device). However, for regulatory scrutiny and to further define the value of triple therapy, it is necessary to provide evidence from a prospective study to answer some of these key questions. We have, therefore, designed a large clinical study, InforMing the PAthway of COPD Treatment (IMPACT), to address these issues.
IMPACT is a phase III, randomised, double-blind, three-arm, parallel-group, global multicentre study designed to prospectively assess the benefits and risks of the closed triple FF/UMEC/VI therapy versus the treatment options of UMEC/VI and FF/VI dual therapies in symptomatic patients with COPD and a history of exacerbations.
Methods
Study design and participants
This is a superiority study, which is designed to show the benefit of FF/UMEC/VI over FF/VI and UMEC/VI. The study aims to randomise approximately 10 000 patients from approximately 1070 centres worldwide. The full study protocol is provided online (www.gsk-clinicalstudyregister.com/study/116855#ps) and the study centres are listed in the online supplementary material.
The study will enrol symptomatic patients with COPD with a forced expiratory volume in 1 s (FEV1) <50% of predicted and a history of one or more moderate or severe (hospitalised) exacerbations within the previous year, or COPD with an FEV1 of 50–80% of the predicted normal and two or more moderate or one severe (hospitalised) exacerbation in the previous year, in line with the GOLD D classification [4]. An overview of the key study inclusion and exclusion criteria is provided in table 1, including allowed and prohibited concomitant medications. The study has been designed to be as permissive as possible with regards to the inclusion of patients with significant cardiovascular disease in order to allow an assessment of safety that is more representative of the targeted population than often seen in clinical trials (e.g. patients with a past history of previous myocardial infarction (>6 months prior to screening) and New York Heart Association Class 1–3 heart failure are eligible, and QTc exclusion criteria have been relaxed, in contrast to the phase III studies on the individual assets and many other similar studies).
Key inclusion and exclusion criteria
Ethical approval and patient consent
The study complies with the Declaration of Helsinki and good clinical practice standards from the International Conference on Harmonisation of technical requirements for the registration of pharmaceuticals for human use, and will be approved by institutional review boards/local ethics committees before study commencement. All patients will have provided written, informed consent before participating in the study.
This study will be guided by an independent data monitoring committee (IDMC), which will monitor the progress of the study and ensure that it meets ethics and patient safety standards. In addition to the IDMC Chair who is a pulmonologist, the IDMC comprises three people (an independent statistician, an independent respiratory clinician with experience in COPD and a cardiologist). Only the IDMC will be authorised to review un-blinded interim safety analyses during the trial, which will be performed and delivered to the IDMC by an independent statistical data analysis centre. In addition, a separate adjudication committee will be established to independently review and categorise the cause of each serious adverse event (SAE) and death in the study. The committee members will remain blinded to treatment.
Randomisation and masking
The study will consist of a 2-week run-in period, a 52-week treatment phase and a 1-week safety follow-up phase. Patients will be randomised 2:2:1 to one of three treatment groups: FF/UMEC/VI 100/62.5/25 µg, FF/VI 100/25 µg or UMEC/VI 62.5/25 µg, respectively, delivered via an identical ELLIPTA dry powder inhaler (GlaxoSmithKline). Patients will be randomised using the proprietary RandAll software (GlaxoSmithKline), and assigned to treatment using the Randomisation and Medication Ordering System (RAMOS; GlaxoSmithKline). Throughout the study, patients will attend seven or eight outpatient clinic visits (figure 1 and table 2): a screening visit, day 1 (randomisation), weeks 4, 16, 28, 40 and 52, plus follow up by telephone 1 week after completion of study treatment. Patients who permanently discontinue study treatment will be encouraged to continue in the study by participating in telephone contacts in order to assess exacerbations, SAEs and concomitant medications post-treatment.

Study design. FF: fluticasone furoate; UMEC: umeclidinium; VI: vilanterol; V: visit; wk: week.
Time of patient visits to the clinic during the study and events planned during each visit
Study endpoints
The study is designed primarily to address the impact on COPD exacerbations, which is reflected in the co-primary endpoints: the annual rate of on-treatment moderate and severe exacerbations comparing FF/UMEC/VI with UMEC/VI and FF/VI. As such, the primary hypothesis to be tested is, the addition of the triple combination therapy, FF/UMEC/VI, will reduce moderate-to-severe acute exacerbations of COPD, compared with either FF/VI or UMEC/VI alone. A number of secondary endpoints will be assessed and the key ones are changes in QoL and lung function. We have also defined, a priori, other secondary endpoints, as well as the corresponding definitions and analysis plans, including incidence of pneumonia and cardiovascular events as safety endpoints and the relative magnitude of benefit of the three therapies on exacerbation prevention by baseline blood eosinophil count. A complete list of study endpoints is provided in table 3. Throughout the study, each COPD exacerbation will be categorised based on severity, as mild, moderate or severe; the definitions of these categories are shown in table 4. Blood samples for eosinophil counts will be taken at the screening visit and at the randomisation visit. This will provide two baseline values, 2 weeks apart, to allow assessment of stability. Further blood samples for eosinophil counts will be taken at weeks 16, 28 and 52, or when the investigational product is discontinued.
Study endpoints
Chronic obstructive pulmonary disease (COPD) severity category definitions
Statistical analyses
For the primary analysis, this study has been designed to have 90% power to detect a 15% reduction in the annual rate of moderate and severe exacerbations with FF/UMEC/VI compared with UMEC/VI, and a 12% reduction compared with FF/VI. A total of 4000 patients will be randomised to receive FF/UMEC/VI, 4000 to receive FF/VI and 2000 to receive UMEC/VI. In addition, the study will have >90% power to detect a 20% reduction in moderate and severe exacerbations for FF/UMEC/VI compared with UMEC/VI, for the subset of patients that has eosinophils ≥150 cells·µL−1.
Sample size calculations assume that the ICS effect will be largely maintained and are based on previous studies. Observed treatment benefits of approximately 20–30% when ICSs were added to LABA, compared with LABA monotherapy [7, 27] powering at a 15% reduction allows for our estimate of the bottom end of the confidence for previously observed reductions. When using a dual bronchodilator, reductions of 10–12% have been observed compared with LAMA monotherapy [17, 19]; therefore, the study has been powered at the upper end of that estimate. For the purpose of sample size calculation, the exacerbation rate for FF/UMEC/VI in this study is estimated to be 0.80, based on a previously observed annual rate of 0.92 for on-treatment moderate and severe exacerbations in “high-risk” patients [5] who received FF/VI in two phase III studies (NCT01009463 and NCT01017952) [7].
Sample size calculations are based on a negative binomial model for exacerbation rate and use a two-sided 1% significance level in order to satisfy regulatory requirements for substantial evidence of efficacy from a single study. The estimate of the dispersion parameter for the negative binomial model is 0.75, which is similar to that seen with fluticasone propionate/salmeterol in the Towards a Revolution in COPD Health (TORCH) study [28] and the dispersion in the FF/VI exacerbation programme [7].
Blinded evaluation of exacerbation rates is planned for this study after 1 year of enrolment or when 5000 patients have been randomised, whichever occurs earlier. This evaluation will allow for adjustment of the sample size in case the blinded exacerbation rate is found to be substantially greater than planned. A subsequent evaluation will be done after approximately 18 months of enrolment or when 7500 patients have been randomised, whichever occurs earlier. Any subsequent change to the planned number of randomised patients will be documented in a protocol amendment.
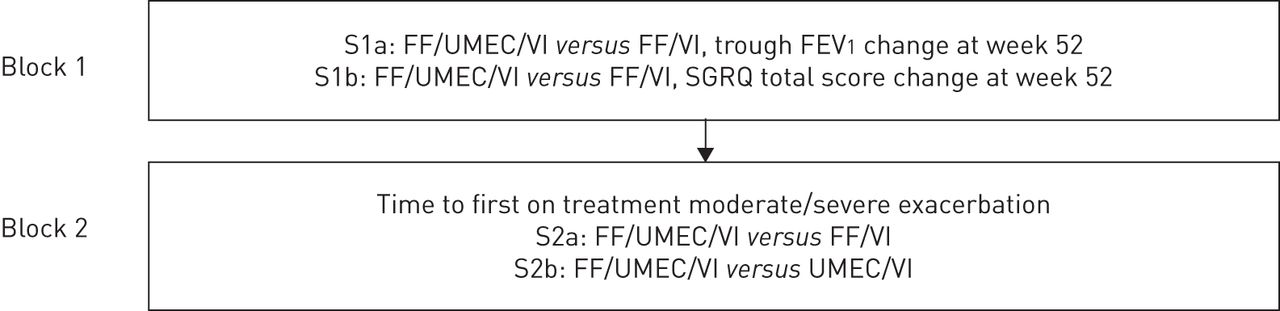
The co-primary endpoints will be controlled for multiplicity using the truncated Hochberg method [29]. A two-sided 5% risk associated with incorrectly rejecting the null hypothesis (significance level) is considered acceptable for this study. The primary comparisons will be declared statistically significant if the p-value for both comparisons is significant at the 0.04 level. If the largest p-value for the two comparisons is above 0.04, the other comparison will be declared statistically significant if the smaller p-value is below 0.025. If at least one of the comparisons is significant, this will allow inference of secondary endpoints. The statistical hierarchy for all secondary endpoints is illustrated in figure 2.
{kind=link}
{kind=link}
Hierarchy of secondary endpoint analyses. S1–2: secondary endpoints; FF: fluticasone furoate; UMEC: umeclidinium; VI: vilanterol; FEV1: forced expiratory volume in 1 s; SGRQ: St George's respiratory questionnaire.
The intent-to-treat (ITT) population will comprise all patients who are randomised to treatment except those randomised in error. This is the primary analysis population and will be used for safety and efficacy analyses. The pharmacokinetic study population will comprise of the subset of patients in the ITT population for whom a pharmacokinetic sample is obtained and analysed; this population will be used to assess all concentration-time and pharmacokinetic parameters.
Discussion
This is the first phase III randomised, double-blind, parallel-group study that will investigate the efficacy and safety of the “closed triple” FF/UMEC/VI therapy. It will assess the relative merits of three different inhaled pharmacological combinations in preventing exacerbations of COPD in symptomatic patients with advanced COPD and a history of exacerbation. Uncertainty lies around the additional benefit of triple therapy including an inhaled steroid over dual bronchodilation without ICSs. This study will address this and will have the power to prospectively answer key questions that may help to identify a more personalised approach towards the use of bronchodilators and ICSs in this patient population. Key secondary aims are to prospectively establish the relationship between blood eosinophils and the benefits of ICS therapy, and to evaluate the relationship between baseline bronchodilator reversibility and the benefits of bronchodilator therapy. Previous attempts have failed to show a consistent intrapatient response and a correlation with outcomes that are not direct measures of airflow limitation (e.g. QoL, exacerbation frequency), likely due to methodological shortcomings. We have hypothesised that, by treating reversibility as a continuous variable and using a modelling approach, we will be able to more accurately describe that relationship.
Historically, the lung function benefit of ICS therapy in patients with more severe COPD has been described as small or modest (consistently shown to be approximately 50 mL) [7]. We will explore if this improvement in lung function in relation to ICS therapy is homogenously spread over the entire population or unequally distributed. If the response is homogenously spread we will determine if it is related to other benefits of ICS therapy, and whether or not it aligns with bronchodilator response to LAMA/LABAs and may modify the same bronchodilator response.
The size of the study will enable characterisation and an accurate evaluation of the impact of any excess pneumonia due to ICS therapy and may also help identify patients at higher risk of pneumonia events. A low eosinophil count at presentation has been associated with pneumonia as one of the few independent markers of poor outcome [30]. However, it is unclear if this relates to a transient marrow suppression associated with acute infection or is linked to the severity of a chronic systemic disease. It has been previously suggested that low body mass index and low FEV1 correlate with the risk of pneumonia and, therefore, the relative risk of excess pneumonia due to ICS therapy [31]. The IMPACT study is large enough to be able to prospectively address these issues.
The IMPACT study does have some limitations. There is the possibility of selection bias as the population is relatively adherent to medication, is able to read and patients with COPD due to exposures other than tobacco are excluded. There is also the possibility that some exacerbations may go unreported, additional data may not be collected due to limitations of the electronic diary and using medication count as a measure of adherence may be inaccurate. Furthermore, while a study duration of 52 weeks is generally accepted as being sufficient for assessing total exacerbations, it may not be enough to draw robust conclusions about severe exacerbations requiring hospitalisation. These will need to be considered when the findings are interpreted.
The strengths of this study include a large, very well-characterised and less restrictive (from a safety perspective) population; the planned characterisation of all suspected pneumonias and moderate/severe COPD exacerbation with a confirmatory radiograph that is independently over-read; the use of an electronic diary to reliably capture symptoms on the day they occur; the provision to collect exacerbation and SAE data in patients who are permanently discontinued from study treatment; and the adjudication of all deaths and SAEs by an independent external adjudication committee.
Conclusion
The recent history of understanding the benefits of inhaled COPD therapy has mostly been driven by large randomised clinical trials such as European Respiratory Society study on chronic obstructive pulmonary disease (EUROSCOP), Inhaled steroids in obstructive lung disease in Europe (ISOLDE), TORCH, UPLIFT, Tiotropium Safety and Performance in Respimat (TIOSPIR), and Study to Understand Mortality and Morbidity in COPD (SUMMIT) [32]. We believe that IMPACT is likely to be another highly important “landmark” study in this population of patients with COPD. It should answer key questions not only about the value of triple therapy compared to dual therapy, but also the relative comparative merits of the two dual therapies (ICS/LABA and LABA/LAMA) and the patient population that would benefit most. As well as prospectively addressing these key questions, we have no doubt that post hoc analyses of the complete data set will generate many more hypotheses ready for testing in future studies. To that end, the individualised patient data will be put into the public domain for the scrutiny of others. More than anything, we are hopeful that this study will make a significant difference to how we understand the therapeutic index of these agents and combinations of agents, leading to improved outcomes for all COPD patients.
Acknowledgements
The authors wish to acknowledge the assistance of the following: David A. Lomas, University College London (London, UK; study design), and Frank Barnhart (protocol preparation and study implementation), Niki Day, Ruby Birk, Michelle Fry, Irene Shupe, Nathalie Richard (all study feasibility and conduct) the late Ann Allen (pharmacokinetics), Maggie Tabberer, Kristina Yu-Isenberg (PRO design), and Sarah Landis (epidemiology input) all of whom were employees of GlaxoSmithKline (London, UK) at the time of protocol finalisation. Editorial support in the form of development of a draft outline, collation of comments and final manuscript submission was provided by Katherine St. John (Gardiner-Caldwell Communications, Glasgow, UK) and Charlotte Kennerley (Gardiner-Caldwell Communications, Macclesfield, UK) and was funded by GlaxoSmithKline.
Author contributions: S Pascoe, D Lipson, N Locantore, I Pavord, H Barnacle, N Brealey and R Mohindra contributed to the study design and drafting the study protocol. R Mohindra helped to operationalise the IDMC and adjudication committees for this study. N Locantore and H Barnacle were involved in the data collection. All authors assisted in the development of the manuscript.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Clinical trial: This study is registered at ClinicalTrials.gov with identifier number NCT02164513.
Support statement: This study was sponsored and funded by GlaxoSmithKline (CTT116855). Funding information for this article has been deposited with the Open Funder Registry.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received December 21, 2015.
- Accepted April 28, 2016.
- Copyright ©ERS 2016
References










