





Abstract
Epidemiological studies have implicated lung inflammation as a risk factor for acute cardiovascular events, but the underlying mechanisms linking lung injury with cardiovascular events are largely unknown.
Our objective was to develop a novel murine model of acute atheromatous plaque rupture related to lung inflammation and to investigate the role of neutrophils in this process.
Lipopolysaccharide (LPS; 3 mg·kg−1) or saline (control) was instilled directly into the lungs of male apolipoprotein E-null C57BL/6J mice following 8 weeks of a Western-type diet. 24 h later, atheromas in the right brachiocephalic trunk were assessed for stability ex vivo using high-resolution optical projection tomography and histology. 68% of LPS-exposed mice developed vulnerable plaques, characterised by intraplaque haemorrhage and thrombus, versus 12% of saline-exposed mice (p=0.0004). Plaque instability was detectable as early as 8 h post-intratracheal LPS instillation, but not with intraperitoneal instillation. Depletion of circulating neutrophils attenuated plaque rupture.
We have established a novel plaque rupture model related to lung injury induced by intratracheal exposure to LPS. In this model, neutrophils play an important role in both lung inflammation and plaque rupture. This model could be useful for screening therapeutic targets to prevent acute vascular events related to lung inflammation.
Abstract
Neutrophils play an important role in atherosclerotic plaque vulnerability related to LPS-induced lung inflammation http://ow.ly/Y7dWW
Introduction
Lung injury and inflammation are considered to be major triggers of acute cardiovascular events [1]. For instance, acute respiratory tract infections and heavy exposures to air pollution increase the risk of ischaemic heart disease, myocardial infarction or stroke by two- to four-fold [2–7]. Most acute cardiovascular events arise from rupture and thrombosis of vulnerable (or unstable) atheromas. Vulnerable atheromas, independently of their size, are characterised by a thin cap, an enlarged “necrotic” core (acellular spaces normally occupied by lipids), increased inflammatory cell infiltration at shoulder regions and areas of haemorrhage resulting from a loss of plaque integrity or leakage of microvessels [1]. In contrast, stable atheromas have a thicker cap, a smaller necrotic core and fewer inflammatory cells. As a result, they are morphologically robust and unlikely to rupture. Although it is generally believed that vulnerable atheromas arise from stable ones, it is not clear how (or even if) acute lung inflammation induces this transition. One major barrier to the investigation of this crucial transition has been the lack of robust animal models that demonstrate atheromatous (plaque) rupture. In small rodents, atheromatous plaques tend to be very stable and rarely demonstrate vulnerability without external manipulation. In this study, we established a novel murine model that uses one-time exposure of lipopolysaccharide (LPS) instilled directly into the lungs via the trachea to promote the transition of stable atheromas to vulnerable ones (inducing plaque rupture) and determined whether circulating neutrophils contribute to this transition.
Materials and methods
Further details are presented in the online supplementary material.
Animal and study design
Male apolipoprotein E-null mice (ApoE−/−) on a C57BL/6J background (stock number: 002052; Jackson Laboratory, Bar Harbor, ME, USA) were fed a Western-type diet containing 21% fat from lard and 0.15% cholesterol (TD88137; Harlan Laboratories Inc., Madison, WI, USA) for 8 weeks to induce atheromatous plaque formation in the brachiocephalic trunk (BCT) [8]. These mice were then randomly divided into three groups: 1) LPS (n=28); 2) saline (n=20); and 3) Western-type diet control (n=6). The experimental scheme is illustrated in figure 1a. In the LPS group, a single dose of 100 µg LPS (Escherichia coli O111:B4; 3 mg·kg−1) (L2630; Sigma-Aldrich, St Louis, MO, USA), dissolved in 50 µL of sterile saline, was sprayed as aerosol particles (16–22 µm in mass median diameter) directly into the lungs through the vocal cords using a microsprayer (MSA-250-M; Penn-Century Inc., Wyndmoor, PA, USA). In the saline group, a single dose of 50 µL sterile saline aerosol was sprayed into the lungs in the same manner [9, 10]. The LPS dosage and timeline were determined based on a pilot study (online supplementary fig. S1a). This study was approved by the animal care committee of the University of British Columbia (A06–1494; A10-0257).

Schematic diagram of main experiments and study designs. a) Experiment 1: atheroma destabilisation by lipopolysaccharide (LPS) instillation in the lungs; b) experiment 2: neutrophil depletion by neutrophil-specific antibody injections; c) experiment 3: early stage of atheroma destabilisation via different routes of LPS exposure. ApoE: apolipoprotein E; WD: Western-type diet; OPT: optical projection tomography; BCT: brachiocephalic trunk; H&E: haematoxylin and eosin; Movat's: Movat's pentachrome staining with Musto/Harrington modification; LPS-ND: LPS-exposed group with neutrophil depletion; LPS-ctrl: LPS-exposed group with control IgG antibody injection; LPS-IT: intratracheal LPS-exposed group; saline-IT: intratracheal saline control group; LPS-IP: intraperitoneal LPS-exposed group; saline-IP: intraperitoneal saline control group. #: n=9 control animals were sacrificed 24 h after first antibody injection without LPS challenge: ND (intraperitoneal injection of anti-Ly6G antibody for neutrophil depletion) n=5; ctrl: intraperitoneal injections of control IgG antibody n=4.
Neutrophil depletion in vivo with antibody injections
To determine the role of neutrophils in plaque rupture in this model, we injected the mice twice intraperitoneally with either 1) rat anti-mouse Ly6G clone 1A8 antibodies (BE0075-1; Bio X Cell, West Lebanon, NH, USA) to deplete circulating neutrophils (LPS-neutrophil depletion (ND) group; n=15); or 2) rat IgG2A isotype clone 2A3 antibodies (BE0089; Bio X Cell) as controls (LPS-ctrl; n=14). The first dose was given 24 h prior to the LPS challenge and the other dose was given immediately after the LPS challenge (fig. 1b). Each dose (400 µg) was diluted in 400 µL sterile PBS [11]. In order to investigate if injections of antibodies on their own induce plaque vulnerability, we included additional comparator groups (without LPS exposure) including 1) mice which were injected intraperitoneally with nonspecific control IgG antibodies as controls (ctrl; n=4) and 2) mice which were injected intraperitoneally with specific anti-Ly6G antibodies, as the neutrophil-specific antibody control group (ND; n=5).
Investigation of early atheromatous destabilisation via intratracheal route versus intraperitoneal route of LPS exposure
To determine the early changes in the plaque, we evaluated plaque morphology at 8 h following 1) intratracheal LPS exposure (LPS-IT; n=16) or 2) intratracheal saline exposure (saline-IT; n=14). To determine whether the route of LPS instillation modified the response in the plaques, two additional groups were used: 1) intraperitoneal LPS exposure (LPS-IP; n=5) and 2) intraperitoneal saline exposure (saline-IP; n=5) [12]. In the LPS-IP group, 100 µg of LPS dissolved in 50 µL of sterile saline was injected directly into the intraperitoneal space. In the saline-IP group, 50 µL of sterile saline was used (fig. 1c for experimental design). The same anaesthetic procedures were used in both IT and IP groups.
Organ and tissue procurement and bronchoalveolar lavage
Under anaesthesia (isoflurane 5% inhalation), blood was collected from the inferior vena cava using a 25-gauge catheter needle (ProtectIV Plus; Smiths Medical International Ltd, Rossendale, UK) 24 h after LPS challenge for cytokine and chemical measurements. Immediate procurement of the heart and blood vessels was performed after blood collection. The heart, aortic arch and branching arteries containing the BCT were carefully harvested and perfused with 3 mL of PBS injected directly into the left ventricle at an infusion rate of 1 mL·min−1 (Harvard Apparatus, Holliston, MA, USA). Bronchoalveolar lavage (BAL) fluid was collected by washing the right lung once with 600 µL of PBS. The left lung was harvested and perfused with 400 µL of 10% formalin at a pressure of 25 cmH2O for histology. Extrapulmonary organs, including liver and kidneys were also harvested and their haematoxylin and eosin (H&E) sections were used for morphological examination.
Optical projection tomography
To visualise the plaques ex vivo without disturbing the plaques, optical projection tomography (OPT) (Bioptonics 3001M; MRC Technology, London, UK) was used, which enabled three-dimensional imaging of the entire aortic arch and BCT of mice [13, 14]. The plaques were assessed in a blinded manner and plaques were considered vulnerable if there was a visible thrombus or evidence for haemorrhage in these plaques (with a size >0.0005 mm3). Thrombi and erythrocytes produced a distinct autofluorescence signal that was 10-fold greater in intensity than the arterial wall, which enabled volume quantification by threshold segmentation. For details of OPT, see the online supplementary material.
Quantitative histology and definition of vulnerable plaque
To correlate the findings of OPT images with histology, the serial cross-sections of the entire BCT, which had been embedded in paraffin (or in optimal cutting temperature compound and frozen in liquid nitrogen) were stained using H&E and Movat's pentachrome stain and examined under the microscope [15]. Vulnerable plaques were defined by the presence of a thrombus or intraplaque haemorrhage. Intraplaque haemorrhage was noted when there were erythrocytes inside the atheroma on at least two contiguous sections spaced 50 µm apart. For details of morphometric analyses, see the online supplementary material and online supplementary fig. S1b and c.
Immunofluorescent staining and signal quantitation
Refer to the online supplementary material for details.
Evaluation of lung inflammation: cell counts in BAL fluid
After centrifugation, the cell pellet in the BAL fluid was resuspended with PBS, gently mixed and cytospun down to a glass slide and fixed with methanol before staining with H&E and Wright–Giemsa stain. Polymorphonuclear (neutrophil) counts were obtained by dividing the cell pellet into four equal quadrants and then taking four random fields from each quadrant using Image Pro software (Media Cybernetics Inc., Rockville, MD, USA) at 400× magnification [16]. Polymorphonuclear leukocytes were morphologically identified by evaluating their size, shape, multinucleation and presence of granules. Monocytes and alveolar macrophages were also determined by morphology. The arithmetic mean of measurements was used for statistical analysis. An automated haemocytometer for total blood cell count was also used to confirm the cell counts in BAL fluid and blood.
Evaluation of systemic inflammation: serum cytokines
Serum samples were used to measure monocyte chemotactic protein-1, keratinocyte chemoattractant, interleukin-6 and tumour necrosis factor-α using Luminex MagPix (EMD Millipore, Billerica, MA, USA) and mouse multiplex assay kit, according to the manufacturers’ protocols.
Plasma LPS measurements and blood chemical test
Plasma concentrations of LPS were measured in duplicate using a commercially available kinetic turbidimetric limulus amebocyte lysate assay kit (Lonza, Walkersville, MD, USA) according to the manufacturer's protocol. Plasma samples were heat-inactivated and then diluted 1:500 in 10 mM magnesium chloride solution to enable positive product control recoveries within 50–200%. Plasma urea and creatinine, lactate dehydrogenase, albumin, alkaline phosphatase, total bilirubin, aspartate aminotransferase, alanine aminotransferase and γ-glutamyl transpeptidase were measured using the ADVIA 1800 Clinical Chemistry System (Siemens, Munich, Germany).
Statistical analysis
All results were expressed as mean±sd, unless otherwise indicated. Data were analysed using Fisher's exact test (for comparison of rupture incidence between two independent groups), t-tests (for comparison of two independent groups that were normally distributed) and Mann–Whitney U-tests (for comparison of two independent groups that were not normally distributed). The Kruskal–Wallis test was used to compare more than two independent groups when distributions of the variables were not normal. All analyses were conducted using Prism 5 (GraphPad Software Inc., La Jolla, CA, USA) and p-values <0.05 (two-tailed) were considered statistically significant.
Results
Atheroma formation in ApoE−/− baseline and LPS-induced inflammation
Morphological measurements of atheroma in the BCT following 8 weeks of a Western-type diet are shown in online supplementary table S1. These mice consistently developed atheromatous plaques in BCT, which were characterised by intimal thickening, foam cell accumulation, a necrotic core and a cap (online supplementary fig. S1b). None of the plaques demonstrated histological features of vulnerability (haemorrhage or thrombus) before LPS or saline exposure. Direct instillation of LPS into the lungs of atherogenic mice resulted in neutrophilic lung inflammation (online supplementary fig. S2a and b) and systemic inflammation characterised by elevated proinflammatory cytokines (online supplementary fig. S3).
Thrombus and intraplaque haemorrhage detected using OPT in LPS-exposed mice
As shown in figure 2, high-resolution OPT revealed the presence of thrombi in two out of three LPS-exposed mice (online supplementary video S1), but none in the three saline-exposed mice (online supplementary video S2). Thrombi were located mostly on the edges of the plaque and extended into the aortic arch (online supplementary video S3). In addition, we detected haemorrhage inside the plaques and the absence of luminal thrombi in one out of three LPS-exposed mice (online supplementary video S4), but none of these phenotypes were observed in saline-treated mice (none out of three mice).

Optical projection tomography (OPT) fluorescence intensity maps and histology of brachiocephalic trunk (BCT). a) Intensity map of BCT from a lipopolysaccharide (LPS)-exposed mouse. Blood and thrombus (red) were determined by their autofluorescence intensity (10 times greater than the vessel wall base intensity). Thrombi were frequently localised at the edges of the vulnerable plaque in LPS-exposed mice. b) Intensity map of BCT from a saline-exposed mouse. No/rare thrombus or intraplaque haemorrhage was observed in the plaques of saline-exposed mice. Scale bars=500 μm. Intensity maps from other LPS-exposed mice, showing c) intraplaque haemorrhage (autofluorescence 10 times greater than the vessel wall); and d) multiple thrombi observed along the aortic arch. The three-dimensional demonstration and navigation of these BCTs are shown in online supplementary videos S1, S2, S3 and S4, respectively. Scale bars=500 μm. e) A two-dimensional (cross-sectional) reconstruction of OPT from a LPS-exposed mouse; f) blood and haemorrhage were observed at the atheroma edge by their high autofluorescent signal and confirmed using histology. Scale bar=100 µm.
Quantitation and characterisation of plaque vulnerability
We quantitated vulnerable plaques using serial histological cross-sections of BCT. This revealed that 17 (68%) out of the 25 LPS-exposed mice harboured vulnerable plaques, which contained histological features of intraplaque haemorrhage or thrombus (fig. 3). Eight (32%) LPS-exposed mice demonstrated luminal blood clot or thrombus, which were mostly located at the edges of the plaque. Nine (36%) LPS-exposed mice displayed haemorrhage inside the atheroma. Plaque complications are summarised in online supplementary figure S4. In contrast, only two (12%) out of the 17 saline-exposed control mice (fig. 3d) demonstrated intraplaque haemorrhage and none of these animals harboured plaques that contained thrombi (p=0.0004 LPS versus saline group).

a) Quantitation of vulnerable plaques in mice. A vulnerable plaque was defined by intraplaque haemorrhage or luminal thrombus in at least two continuous cross-sections (50-μm intervals) stained with haematoxylin and eosin (H&E) and Movat's pentachrome stain. b) A representative brachiocephalic trunk (BCT) cross-sectional image stained using Movat's pentachrome from a lipopolysaccharide (LPS)-exposed mouse. A thrombus was present in the lumen of the BCT at the edge region of the plaque, and loss of endothelium was observed at the adjacent edge of atheroma. c) The representative H&E-stained cross-sectional image of the BCT from the same LPS-exposed mouse. Fibrin deposition around the thrombus and loss of endothelium at the edges of the plaque were observed. d) A representative H&E cross-section of an atheroma edge from a saline-exposed animal. Endothelium integrity was preserved; no thrombi were detected in any saline-exposed animals. Scale bars=100 μm.
The morphometric data of atheroma are shown in table 1. The mean cap thickness (relative to plaque thickness) in the LPS-exposed mice was approximately one-third that in the saline-exposed mice (13±10% versus 39±26%, p=0.0004). The size of the necrotic core in LPS-exposed mice was twice that in the saline-exposed mice (36.9±12.9% versus 18.6±11.8%, p=0.0004). However, the overall plaque size of the LPS-exposed mice (101±41×103 µm2) was not significantly different than that of the saline-exposed mice (78±34×103 μm2, p=0.102).
Morphometry of atheroma using the frozen cross-sections of the brachiocephalic trunk that contained the maximal plaque size
Neutrophil depletion and characterisation of plaque vulnerability
We characterised the vulnerable plaques in the BCT of LPS-ND and LPS-ctrl groups using both histology and OPT (fig. 4). Altogether, 10 (71.4%) out of the 14 LPS-ctrl mice harboured vulnerable plaques, which contained histological features of intraplaque haemorrhage or thrombus. In contrast, only four (26.7%) out of the 15 LPS-ND mice harboured vulnerable plaques (p=0.0268) (fig. 5a). Further plaque characterisations are shown in online supplementary figure S5a and b. Five (35.7%) out of the 14 LPS-ctrl mice demonstrated luminal blood clot or thrombus. Similar to previous findings, these complications were mostly located at the edges of the plaque (online supplementary fig. S5c). Eight (57.1%) out of the 14 LPS-ctrl mice displayed haemorrhage inside the atheroma. In contrast, only two (13.3%) out of the 15 LPS-ND mice demonstrated intraplaque haemorrhage (p=0.0209) and only two (13.3%) of these animals harboured plaques that contained blood clots. The representative BCT images from LPS-ctrl and LPS-ND mice are demonstrated in online supplementary video S5; the representative plaque histology is demonstrated in figure 5b. LPS-exposed mice treated with neutrophil-specific antibodies had reduced plaque vulnerability detected by OPT (LPS-ND versus LPS-ctrl, p=0.0042) (fig. 5c). Without LPS challenge, the injections of nonspecific control IgG antibodies and neutrophil-specific antibodies did not induce plaque vulnerability or change the plaque phenotype (zero out of four and five plaques ruptured, respectively) (online supplementary figure S5d). Immunofluorescent staining of myeloperoxidase (MPO) showed that MPO+ polymorphonuclear cells were localised at the edges and in the core area of plaques in the LPS-ctrl mice, but were markedly reduced in the plaques of LPS-ND mice (fig. 5d and online supplementary fig. S5e–g).

Optical projection tomography (OPT) fluorescence intensity maps of brachiocephalic trunk (BCT). Intensity maps of BCT from a lipopolysaccharide (LPS)-exposed mouse treated with a) neutrophil-specific antibodies (LPS-ND) and b) control antibodies (LPS-ctrl). Blood and thrombi (red) were determined by their autofluorescence intensity (10 times greater than the vessel wall base intensity). Thrombi were frequently localised at the edges of the vulnerable plaque in LPS-ctrl mice. No or rare thrombus or intraplaque haemorrhage was observed in the plaques of LPS-ND mice; the three-dimensional demonstration and navigation of these BCTs are shown together in online supplementary video S5. Intensity maps from antibody control mice: a BCT from a mouse without LPS exposure injected with c) neutrophil-specific antibodies and d) control antibodies. No plaque vulnerability was observed in these groups. Scale bars=500 µm.

a) Quantitation of vulnerable plaques in mice. A vulnerable plaque was defined by the presence of intraplaque haemorrhage or luminal thrombus in the brachiocephalic trunk (BCT) based on histology and optical projection tomography (OPT). Haematoxylin and eosin-stained cross-sections of BCT from b) lipopolysaccharide-exposed mice with neutrophil depletion (LPS-ND) and c) LPS-exposed mice with control IgG antibody injections (LPS-ctrl). Intraplaque haemorrhages (arrows) were observed in the BCT of LPS-ctrl mice. d) Quantitation of vulnerability signals in the BCT detected using OPT. Data are presented as mean±sem. Immunofluorescence-stained cross-sections of myeloperoxidase (MPO; green), a general maker for peripheral neutrophils (blue: nuclear): e) MPO+ neutrophils in the atheroma of LPS-ND mice were markedly reduced by injections of neutrophil-specific antibodies; f) Neutrophil/MPO signal (arrow) was located at plaque edges and in the core area in LPS-ctrl mice. Scale bars=100 µm. g) Composite image of MPO+ polymorphonuclear cell at high magnification. Scale bar=10 µm.
Neutrophil recruitment in the lungs and circulating white blood cell counts
In the absence of LPS exposure, injections of nonspecific control IgG antibodies or neutrophil-specific antibodies into mice did not induce neutrophil infiltrations in their lungs (online supplementary fig. S6a). In the presence of LPS, neutrophil lung infiltrations were observed; neutrophil counts in the BAL were consistently elevated in the LPS and LPS-ctrl group (online supplementary figure S2). However, it was significantly attenuated by the injections of neutrophil-specific antibodies (LPS-ND group versus LPS-ctrl group, p=0.0002). Macrophage number in the BAL remained consistent among the antibody control groups in both the absence and presence of LPS (online supplementary fig. S6b). Administration of neutrophil-specific antibodies significantly reduced the circulating neutrophil counts in the presence of LPS (LPS-ND group 0.10±0.06 versus LPS-ctrl group 0.8±0.5×109·L−1; Mann–Whitney test p=0.0021). All lymphocyte and monocyte counts in the blood were not significantly different between LPS-ND and LPS-ctrl groups, or between ND and ctrl groups. The white blood cell counts of the various groups are shown in online supplementary table S2.
Early stage of plaque destabilisation at 8 h via intratracheal but not intraperitoneal route of LPS instillation
To investigate the earlier stages of atheromatous destabilisation, we examined plaques in BCT at 8 h post-intratracheal LPS instillation using a dissection microscope and OPT. At 8 h, there was evidence for plaque vulnerability in BCT (LPS-IT: 8/16 versus saline-IT: 1/14, p=0.017). As shown in figure 6a and b, thrombus and haemorrhage were detectable in the BCT. The vulnerability signals of randomly selected BCT (11 LPS-IT mice versus 10 saline-IT mice) were quantitated. The LPS-IT group demonstrated a significantly greater signal than the saline-IT group (p=0.04). We then investigated whether or not similar changes could be found by injecting the same LPS dose intraperitoneally. There was no significant difference in the occurrence of vulnerable plaques between intraperitoneal LPS instillation and saline controls (LPS-IP: 1/5 versus saline-IP 0/5; p=1.0). Both LPS groups (IT and IP) demonstrated higher plasma LPS levels compared with the saline control groups. Interestingly, plasma LPS levels increased 17 000-fold when LPS was injected intraperitoneally, but the same dose given intratracheally increased the plasma LPS levels by only 40-fold (fig. 6c). Mice injected with intraperitoneal LPS also demonstrated higher plasma lactate dehydrogenase and urea levels compared with mice exposed to intratracheal LPS (fig. 6d). The BAL cell count data showed that neutrophil and macrophage counts were not significantly different between the LPS-IP and saline-IP groups; however, the LPS-IT group demonstrated higher neutrophil counts (online supplementary fig. S6c and d). Circulating white blood cell counts revealed very similar patterns between the intratracheal route and the intraperitoneal route of LPS exposure (online supplementary fig. S7). Together, these data suggest that intratracheal administration of LPS induced neutrophilic lung inflammation, but resulted in less systemic absorption and reduced injury to the liver and kidneys compared with intraperitoneal administration of LPS. Importantly, intratracheal LPS instillation (but not intraperitoneal injection) promoted transition of stable plaques to vulnerable ones in this model.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
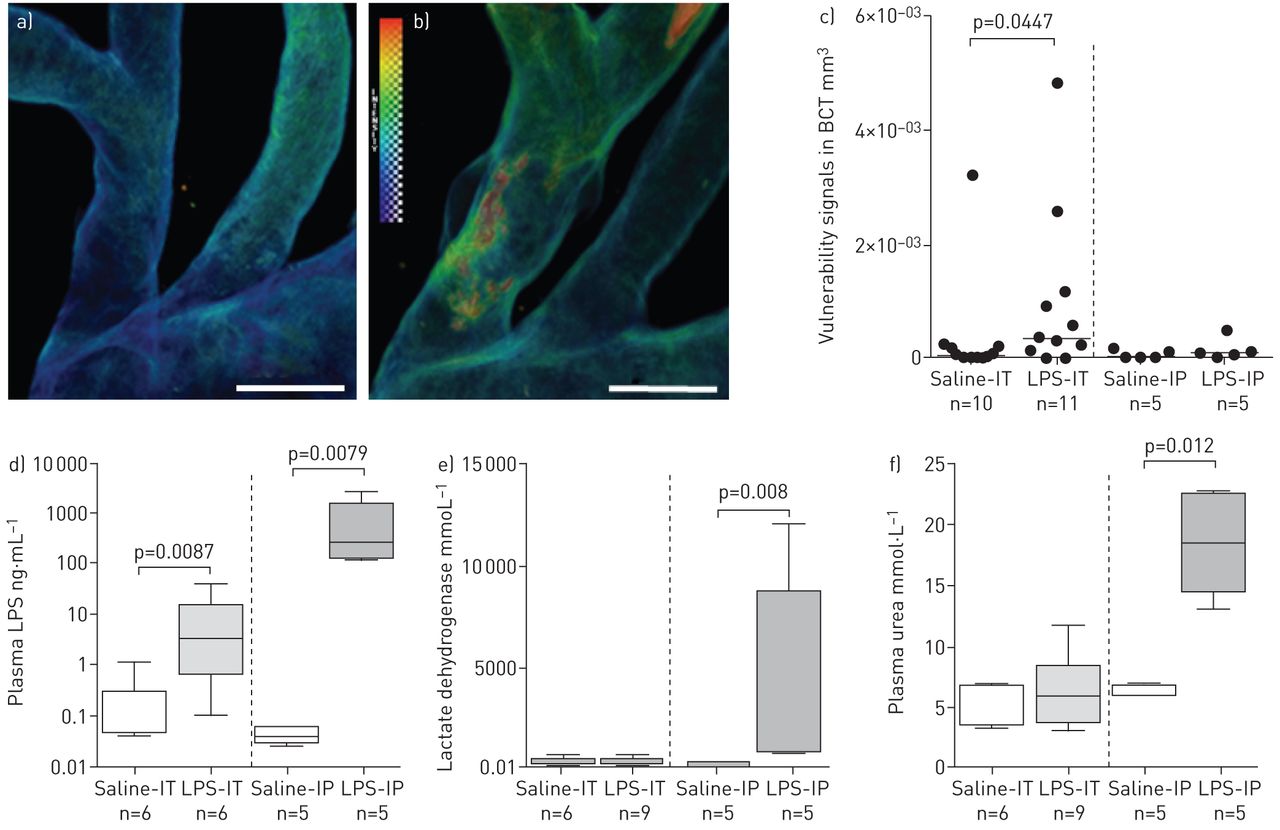{kind=link}
Early stages of plaque destabilisation, lipopolysaccharide (LPS) translocation and tissue damage at 8 h time point via different routes of LPS exposure. Representative optical projection tomography intensity map of the brachiocephalic trunk (BCT) from a) a control mouse instilled intratracheally with saline (saline-IT) and b) an LPS-exposed (intratracheal administration) mouse (LPS-IT). Vulnerability signals are shown in red. Scale bars=500 µm. c) Quantitation of vulnerability signals in the BCT. The median signal intensity (in volume) of the LPS-IT group was higher than that of the saline-IT group (p=0.04). >45% of LPS-IT mice demonstrated signs of destabilisation, but these signs only occurred in 10% of saline-IT saline controls; the median signal intensity of the intraperitoneal LPS-exposed mice (LPS-IP) was no different from that of the intraperitoneal saline control (saline-IP) group. There was no increase in plaque vulnerability between the LPS-IP and saline-IP groups. Bars represent medians. d) Plasma LPS level in LPS-IT group was slightly (<40-fold) higher than that in saline-IT group. Plasma LPS levels in the LPS-IP group were markedly higher (17 000-fold) than that in the saline-IP group. e) Lactate dehydrogenase in the plasma following different routes of saline or LPS exposure. Elevation of plasma lactate dehydrogenase was observed in the LPS-IP group when compared to the saline-IP group. Plasma lactate dehydrogenase is commonly used as an indicator for systemic tissue damage (muscles, liver, heart, etc.). f) Urea level in the plasma following exposure to LPS and saline via different routes. Plasma urea is a common marker of kidney damage. It was elevated in the LPS-IP group, but not the LPS-IT group.
Discussion
We found that a single intratracheal administration of LPS caused atheromatous plaques to become vulnerable within 24 h of exposure. By 8 h post-instillation, plaques demonstrated features of vulnerability including haemorrhage and thrombus formation, which could be detected and quantified using OPT. However, administration of the same dose of LPS intraperitoneally failed to induce these acute changes, despite causing greater rises in serum LPS levels and more end-organ damage in liver and kidneys. These findings highlight the importance of lung inflammation and injury in the pathogenesis of acute plaque rupture. Additionally, we found that selective depletion of circulating neutrophils prior to intratracheal LPS exposure prevented plaque vulnerability and rupture, suggesting that neutrophils play a key role in the pathogenesis of plaque vulnerability induced by lung injury. Our findings are consistent with the concept of neutrophilic inflammation in the arteries during acute cardiovascular events [17]. It is postulated that during cardiovascular events, neutrophils are activated and recruited to the coronary arteries where they release azurophilic (primary) granules, amplifying the original inflammatory signal. In this study we have experimentally validated the importance of neutrophils in plaque rupture related to lung inflammation due to acute LPS exposure.
Our study has several strengths. It is widely believed that one of the very early plaque changes leading to acute cardiovascular events is the transition of stable plaques to vulnerable ones, characterised by haemorrhage followed by rupture and then thrombosis. However, the mechanism(s) by which this sequence of events occurs are largely unknown. A major barrier to mechanistic studies and therapeutic discoveries in this field has been the lack of a simple animal model of plaque rupture. To address this critical gap, previous studies have used a variety of interventional approaches using surgery (for example by creating tandem stenosis to arteries [18]) or drugs (e.g. angiotensin II infusions directly into plaque-containing arteries [19]) or prolonged feeding with a high-fat diet [8]. In this experiment, unlike previous studies, which focused on chronic factors, we exploited the strong epidemiological association between lung injury and acute cardiovascular events and demonstrated that a single dose of intratracheal (but not intraperitoneal) LPS can consistently cause stable plaques to become vulnerable and induce acute plaque rupture. Furthermore, unlike to previous studies, which solely relied on histology to demonstrate plaque rupture, we used OPT, a novel ex vivo micro-imaging tool that does not require tissue sectioning and thus preserves the architecture of the vessel. This technique enables high-resolution, three-dimensional imaging of organs/tissues. It can also be combined with whole-mount immunofluorescent staining technology for studying the distribution of specific molecules in organs and tissues. By combining this new technique with our histological findings, we provide convincing evidence that acute lung injury induces plaque rupture. This novel model can now be used to determine mechanistic pathways and, most importantly, test novel compounds to prevent acute plaque rupture related to acute lung inflammation.
We have used LPS as a convenient trigger to induce lung injury and inflammation, simulating an acute lung infection with Gram-negative bacteria. Future studies should explore more common human pathogens, such as respiratory viruses or bacteria, which would enhance the application and generalisability of this model to clinical conditions. However, the trade-off could be a more complicated model of lung inflammation and host responses that may be difficult to characterise fully. Interestingly, LPS administered intraperitoneally did not induce plaque rupture, suggesting that the systemic inflammatory response induced by lung injury may be unique with regard to its downstream effect on blood vessels. This concept needs further evaluation.
In this model we showed that circulating neutrophils contribute to the LPS-induced acute destabilisation and rupture of plaques. It was beyond the purview of this study to evaluate downstream molecules directly responsible for plaque rupture. Based on some preliminary experiments, we speculate that MPO and metalloproteinases could be important downstream factors in this process (online supplementary fig. S4f) [20–30], but additional studies are needed to determine the exact molecular pathways that lead to acute plaque destabilisation and rupture. It should also be noted that we did not identify the mediators responsible for the activation and recruitment of neutrophils into the plaques following LPS stimulation. Further work will be needed to unravel the molecular underpinnings of this process.
Notwithstanding these limitations, this study has established a new mouse model in which lung exposure to LPS leads to lung inflammation and causes acute plaque destabilisation. Furthermore, we showed that in this model neutrophils contribute to plaque destabilisation. This model, particularly when coupled with the OPT imaging technique, could be a useful tool to interrogate possible therapeutic targets to prevent acute plaque destabilisation and thereby reduce the burden of cardiovascular morbidity and mortality related to lung injury [31].
Acknowledgements
We thank Wang Xi (Center for Heart Lung Innovation, University of British Columbia and St Paul's Hospital, Vancouver, Canada) for technical assistance in the animal experiments and David Granville (Center for Heart Lung Innovation, Dept of Pathology and Laboratory Medicine, University of British Columbia and St Paul's Hospital), Pascal Bernatchez (Center for Heart Lung Innovation, Dept of Anesthesiology, Pharmacology and Therapeutics, University of British Columbia and St Paul's Hospital) and Hye-Yun Park (Division of Pulmonary and Critical Care Medicine, Dept of Medicine, Samsung Medical Center, Sungkuynkwang University, Seoul, South Korea) for useful feedback and advice. We thank Nicola Bai and Alon Hendel for providing helpful comments on morphometry, Claire Smits and her Genetically Engineered Models team for assistance in preparing ethics applications and equipment arrangement, Amrit Samra for assisting with tissue processing and Rachel Chen for assistance with immunofluorescent imaging (Center for Heart Lung Innovation, University of British Columbia and St Paul's Hospital).
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This work was supported by the Canada Research Chairs Program and the Canadian Institutes of Health Research. D.D. Sin is a tier 1 Canada Research Chair in chronic obstructive pulmonary disease. Optical projection tomography was performed in the Centre for High-Throughput Phenogenomics at the University of British Columbia (UBC), a facility supported by the Canada Foundation for Innovation, British Columbia Knowledge Development Foundation, and UBC Faculty of Dentistry.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received June 19, 2015.
- Accepted January 25, 2016.
- Copyright ©ERS 2016
References










