





Abstract
Obstructive sleep apnoea (OSA) has been inconsistently associated with insulin resistance and adverse metabolic states. We aimed to assess independent contributions of OSA to insulin resistance and dyslipidaemia in a large paediatric cohort.
Habitually snoring children underwent overnight polysomnography, anthropometric measurements and fasting laboratory evaluations. Primary outcome measures included insulin, glucose, homeostasis model of insulin resistance, lipoproteins and sleep disturbance measures.
Among 459 children aged 5–12 years, obesity was the primary driver of most associations between OSA and metabolic measures, but sleep duration was inversely associated with glucose levels, with N3 and rapid eye movement (REM) sleep being negatively associated and sleep fragmentation positively associated with insulin resistance measures. In children with mild OSA, the presence of obesity increased the odds for insulin resistance, while higher apnoea/hypopnoea index values emerged among obese children who were more insulin-resistant.
The exclusive presence of interactions between OSA and obesity in the degree of insulin resistance is coupled with synergistic contributions by sleep fragmentation to insulin resistance in the context of obesity. Insufficient N3 or REM sleep may also contribute to higher glycaemia independently of obesity. Studies are needed to better delineate the roles of puberty and sleep fragmentation in insulin resistance and the metabolic syndrome.
Abstract
OSA and obesity jointly affect insulin resistance, but insulin resistance is absent in nonobese children with OSA http://ow.ly/W4BmY
Introduction
Paediatric obesity prevalence has increased in recent decades [1], with a concomitant increase in obesity-related comorbidities, including type 2 diabetes mellitus (T2DM) [2] and the metabolic syndrome [3]. While weight loss can reduce the risk of developing obesity-related comorbidities [4], many find weight loss maintenance difficult [5]. Thus, it is imperative to identify other modifiable risk factors.
One potential candidate for intervention is obstructive sleep apnoea (OSA) syndrome, a common comorbidity of obesity [6] characterised by repetitive upper airway collapse during sleep, oxyhaemoglobin desaturation and recurrent arousals from sleep, i.e. sleep fragmentation [7]. The metabolic effects of paediatric OSA have been previously explored only in a relatively restricted number of studies, with somewhat inconsistent findings. In some studies, paediatric OSA has been shown to associate with visceral adiposity [8] and insulin resistance [9, 10]; however, another study found that insulin resistance was primarily determined by obesity, not OSA [11]. Given these contradictory results, we set out to prospectively study a large cohort of snoring children, and examine the independent contribution of OSA to insulin resistance and dyslipidaemia.
Materials and methods
Subjects
This was an observational study of healthy snoring children, approved by the University of Louisville Human Research Committee (protocol 474.99) and the University of Chicago Institutional Review Board (protocol 09-115-B). Snoring children aged 5–12 years undergoing evaluation for suspected OSA were prospectively recruited. Children with genetic syndromes or chronic medical conditions (excepting well-controlled asthma on no controller medications) and children receiving medications potentially affecting sleep, insulin or glucose homeostasis, or lipids (e.g. systemic glucocorticoids within 1 month of the study) were excluded from participation. Informed consent and age-appropriate assent were obtained.
Anthropometric measurements
All participants underwent anthropometric measurements. An electronic scale assessed weight. Height was assessed using a wall-mounted stadiometer. Body mass index (BMI; kg·m−2) was calculated. Age- and sex-adjusted standard scores (z-scores) for BMI were calculated [12]. Study participants with BMI z-score ≥1.64 (BMI ≥95th percentile) were deemed obese [13]; other children were classified as nonobese.
Overnight polysomnography
All participants underwent overnight polysomnography (PSG) at Kosair Children's Hospital (Louisville, KY, USA) or at the Pediatric Sleep Laboratory at the University of Chicago Medical Center (Chicago, IL, USA). Parameters recorded and digitised using commercially available software included: eight-channel electroencephalogram (EEG), chin and bilateral anterior tibial and forearm electromyograms, bilateral electro-oculogram, heart rate by ECG, chest and abdominal wall movement by semicalibrated respiratory inductance plethysmography with derivation of sum from Lisajou analysis, airflow monitoring via side-stream end-tidal capnography, breath-by-breath assessment of end-tidal carbon dioxide levels via capnography (BCi International, Waukesha, WI, USA), nasal pressure transducer (Braebon, Kanata, ON, Canada), oronasal thermistor, pulse oximetry (SET; Massimo, Irvine, CA, USA) with simultaneous recording of the pulse waveform for arterial oxygen saturation (SpO2), and analogue output from a body position sensor were simultaneously monitored. All measures were digitised using a commercially available PSG system (Nihon Kohden, Tokyo, Japan). Sleep architecture, respiratory events and arousals were scored by the same investigators (L.K.G., R.B. and D.G.) serially at the two sites using standard paediatric criteria [14].
Apnoeas were defined as ≥90% decrement in airflow lasting at least two breaths and hypopnoeas were defined as >50% decrement in nasal airflow accompanied by either a 3% desaturation or an EEG arousal from sleep lasting ≥3 s. Arousals were defined as per the revised American Academy of Sleep medicine guidelines [14]. Mean and nadir SpO2 were assessed. Children with an obstructive apnoea/hypopnoea index (AHI), defined as number of apnoeas and hypopnoea per hour of sleep, of <1.0 events·h–1 total sleep time (TST) were deemed to have normal breathing during sleep, children with AHI 1–5 events·h–1 TST were considered to have mild OSA, and children with AHI ≥5 events·h–1 TST were considered to have moderate–severe OSA [11].
Metabolic parameters
The morning following the sleep study, participants had fasting blood drawn. Fasting plasma insulin (FPI) levels were measured using a radioimmunoassay kit (Coat-A-Count Insulin; Diagnostics Products, now Siemens Medical Solutions Diagnostics, assay no longer manufactured), sensitivity 1.2 μIU·mL−1, and subsequently using a solid-phase, two-site chemiluminescent immunometric assay (Immulite 2000; Siemens Medical Solutions Diagnostics, Los Angeles, CA, USA), measuring range 2–300 μIU·mL−1. Of note, the initial insulin assay was more sensitive, allowing insulin values between 1.2 and 2 μIU·mL−1 to be determined; children with equivalent insulin sensitivity with insulin measured using the second assay would have insulin values reported as <2 μIU·mL−1, meaning homeostasis model assessment of insulin resistance (HOMA-IR) and the McAuley index could not be calculated or mean HOMA-IR values for the group measured with the second insulin assay. To avoid introducing a potential bias between children whose insulin levels were measured with the two different methodologies due to measurement sensitivity differences, any child with FPI <2.0 μIU·mL−1 was excluded from analyses. Fasting plasma glucose (FPG) was measured using a hexokinase glucose 6-phosphate dehydrogenase method (Flex Reagent Cartridges; Dade Behring, Newark, DE, USA) and using a UV enzymatic method with hexokinase (Roche Cobas 8000 702 platform; Roche, Basel, Switzerland); FPG levels <63 mg·dL−1 were excluded from analyses as artefactual (likely due to excessive specimen processing time). Children with FPG values consistent with T2DM (>125 mg·dL−1) were excluded from analyses, as they constitute a separate metabolic disease population.
High-density lipoprotein (HDL), low-density lipoprotein (LDL), total cholesterol and triglycerides were measured using homogenous enzymatic colorimetric assays (Roche Cobas 8000 502 platform). Analytical measuring range and assay coefficients of variations (CVs) were: 1) total cholesterol: range 3.86–800 mg·dL−1, CV ≤1.6%; 2) HDL cholesterol: range 3–120 mg·dL−1, CV ≤1.5%; 3) LDL cholesterol: range 3.86–548 mg·dL−1, range ≤2.7%; and 4) triglycerides: range 8.85–885 mg·dL−1, CV ≤2.0%. Total cholesterol/HDL, LDL/HDL and triglyceride/HDL ratios were calculated. Higher values for total cholesterol/HDL [15], LDL/HDL [16] and triglyceride/HDL [17] ratios are associated with increased cardiovascular disease risk.
Calculated insulin sensitivity parameters
HOMA-IR was calculated as: (FPI (μIU·mL−1)×FPG (mg·dL−1))/405 [18, 19]. HOMA-IR values >2.5 were considered “abnormal” (i.e. consistent with insulin resistance) [20].
The McAuley index, which incorporates a weighted combination of FPI and triglycerides [21], was calculated as: exp(2.63–0.28×ln(FPI (μIU·mL−1)–0.31×ln(triglycerides (mmol·L−1)). Lower values denote greater insulin resistance.
Statistical analyses
Statistical analyses were performed using SPSS version 22.0 (IBM, Armonk, NY, USA). Skewed variables were natural log-transformed to normalise distributions. Correlation analyses were used to examine associations between OSA measures and anthropometric and metabolic variables. Stepwise multivariate linear regression models were constructed to assess relationships between sleep measures (e.g. TST or AHI) and metabolic variables (e.g. FPI) while controlling for BMI and, if significantly associated on correlation analyses, age. A p-value<0.05 was used as the cut-off for statistical significance. No differences emerged between the two study sites; as such, the data were merged and the cohort analysed as one group. As obese participants could be more susceptible to OSA's metabolic sequelae, and as severe versus mild OSA could have a differential metabolic impact, subjects were divided into three groups of OSA severity, and subdivided into nonobese and obese within OSA groups. Chi-squared tests were used to compare categorical characteristics, while ANOVA or Kruskal–Wallis tests, as appropriate, were used to compare continuous metabolic and sleep characteristics among the groups.
To further assess the impact of OSA upon insulin resistance, we compared sleep measures among children at extremes of insulin sensitivity. Children with intermediate insulin sensitivity were excluded from this stage of the analysis in order to see whether more subtle differences in sleep architecture and sleep disturbances between children with greatest and lowest insulin sensitivity would emerge. HOMA-IR quartiles were calculated for the entire study population and children in the lowest and highest quartiles (quartile 1, HOMA-IR <0.91; quartile 4, HOMA-IR >2.33) within each AHI and BMI group were included in a further subanalysis. t-tests or Mann–Whitney U-tests were performed to compare differences in OSA elements, age and degree of obesity between children in the lowest versus highest HOMA-IR quartiles within each subgroup.
Results
Study population
537 children completed the study. Of these, two children were excluded for FPG >125 mg·dL−1, 13 were excluded for FPG <63 mg·dL−1 and 62 were excluded for insulin levels <2 μIU·mL−1. Thus, 460 children were included in the final cohort: mean age 7.2±1.2 years, 58% male, 167 obese, 59% Caucasian, 33% African-American, 6% Hispanic and 2% of other racial or ethnic origins. Baseline demographic characteristics by subgroup are shown in table 1. Demographic comparisons showed no significant differences in age, sex, race/ethnicity or height across OSA groups, or between nonobese and obese participants within OSA groups. BMI and BMI z-score differed significantly only in obese children across AHI groups, with highest values seen in moderate–severe OSA. 53% of all study participants had OSA and 17% had moderate–severe OSA (AHI >5 events·h–1 TST). The prevalence and severity of OSA were higher in nonobese versus obese participants; 65% of obese participants had some degree of OSA and 31% had moderate–severe OSA, while 46% of nonobese participants had some degree of OSA and only 9% had moderate–severe OSA.
Demographics and anthropometrics: comparisons between nonobese and obese apnoea/hypopnoea index (AHI) groups
Metabolic parameters
Age was positively associated with BMI (correlation coefficient (p-value) 0.207 (<0.0005), BMI z-score (0.099 (0.035)), FPI (0.111 (0.018)) and HOMA-IR (0.116 (0.013)), and negatively with the McAuley index (−0.113 (0.016)). Similarly, BMI was positively associated with FPI (0.093 (0.045)), HOMA-IR (0.099 (0.033)) and triglycerides (0.096 (0.041)), and negatively with the McAuley index (0.124 (0.0008)), while BMI z-score was significantly negatively associated with the McAuley index only (−0.091 (0.050)); glucose levels trended to associate positively with age and BMI, while remaining lipoprotein levels were not associated with age, BMI or BMI z-score (data not shown). These results showed that older and more obese children were more likely to be insulin resistant.
Correlation analyses examining associations between sleep variables and anthropometric and metabolic outcomes are shown in tables 2 and 3 and, for lipids, in supplementary table S1. AHI, apnoea index, obstructive apnoea index and all arousal indices (ArIs) were significantly positively associated with BMI and BMI z-score (spontaneous and total ArIs only were associated with age), and nadir and mean SpO2 associated negatively with BMI and BMI z-score, i.e. more severe OSA accompanied greater obesity. The spontaneous and total ArIs were associated positively with FPI and HOMA-IR, and negatively with the McAuley index, suggesting greater insulin resistance in those with more fragmented sleep; the total ArI also associated positively with FPG and triglycerides. FPG, FPI and lipoprotein levels were not associated with apnoea indices or SpO2. In support of these findings, the proportions of rapid eye movement (REM) sleep and delta wave sleep (stage N3) were also significantly but modestly associated with FPI; percentage TST in REM also associated modestly with HOMA-IR. TST was strongly negatively associated with FPG (as well as with age, BMI and BMI z-score). Percentage TST in N3 was significantly negatively associated with FPG. No other sleep architecture measure associated significantly with any metabolic outcome examined.
Associations between obstructive sleep apnoea measures and anthropometric and metabolic parameters in entire study population
Associations between sleep architecture and anthropometric and metabolic parameters in entire study population
Comparisons of metabolic measures between nonobese and obese children within OSA groups and among nonobese and obese across OSA groups are depicted in table 4 and supplementary table S2. No significant metabolic differences were seen between either nonobese children or obese children with varying degrees of OSA. The McAuley index was significantly lower in obese versus nonobese children with moderate–severe OSA, indicating greater insulin resistance in the obese population; obese children without OSA and with mild OSA demonstrated no significant metabolic differences compared with nonobese children, although trends were seen towards lower higher FPI and triglycerides in obese versus nonobese children with mild OSA (figure 1).
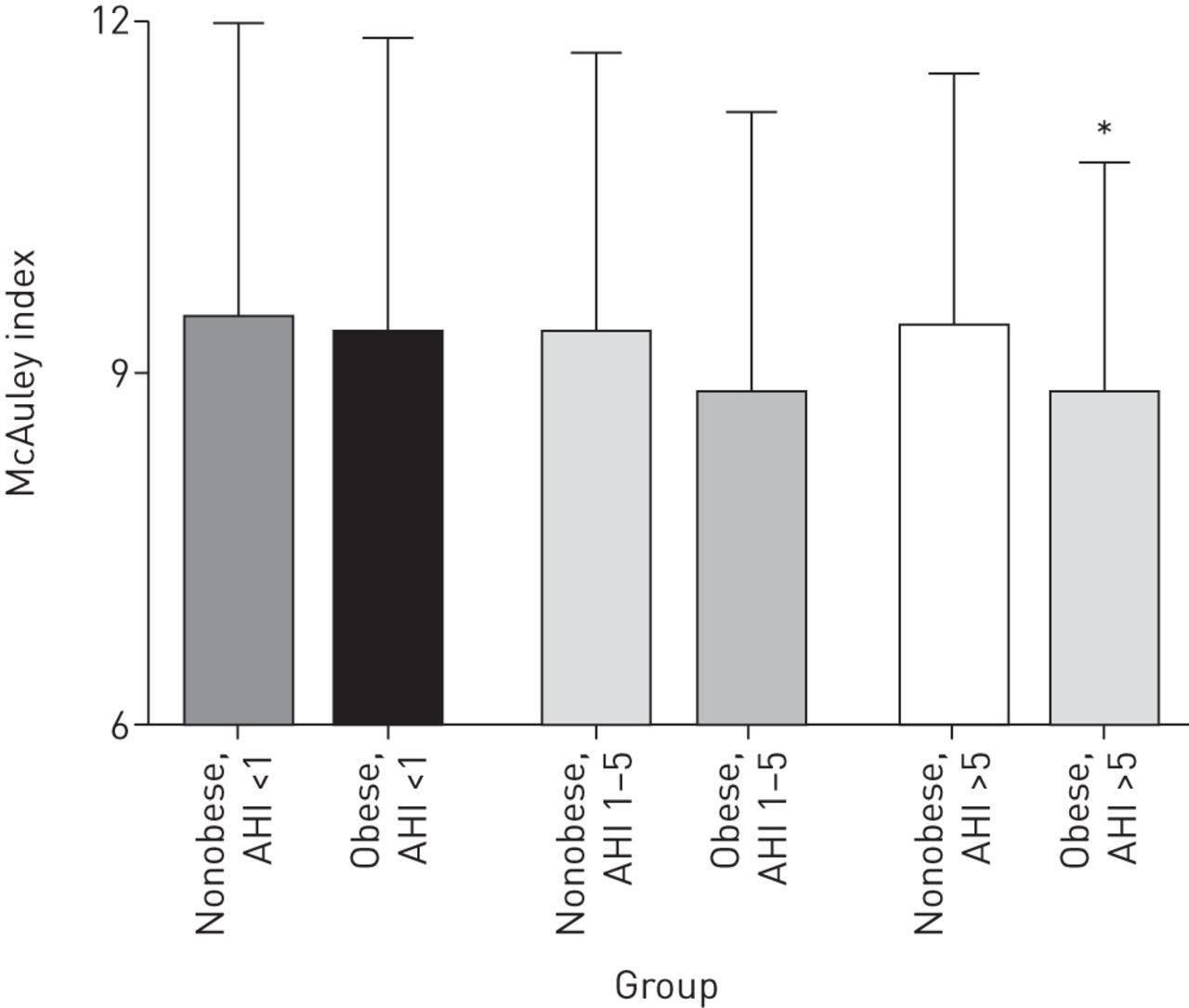
McAuley index. Data represent mean±sd. Comparisons were made across the groups. *: p<0.05 for comparison between nonobese and obese participants with the apnoea/hypopnoea index (AHI; events·h–1) group.
Comparison of metabolic parameters across nonobese and obese apnoea/hypopnoea index (AHI) groups
The distribution of OSA measures is depicted in table 5. AHI differed among the groups by definition. Comparing sleep parameters across nonobese and across obese children with varying degrees of OSA, those with moderate–severe OSA had significantly lower SpO2 values, more sleep fragmentation due to respiratory events and higher sleep pressure score (a surrogate measure of sleep fragmentation [22]). Comparing sleep disturbance measures between nonobese and obese children within OSA subgroups, we found no differences between nonobese and obese children without OSA, lower mean and nadir SpO2 levels in obese versus nonobese children with mild OSA, and significantly higher AHI, respiratory ArI, and lower mean and nadir SpO2 in obese versus nonobese children. The differences in mean AHI (19.4 versus 11.2) and nadir SpO2 (79.1 versus 87.4%) were also of considerable clinical significance. In other words, even within the moderate–severe OSA group, the obese children had greater disease severity than the nonobese children.
Obstructive sleep apnoea (OSA) elements results across nonobese and obese apnoea/hypopnoea index (AHI) groups
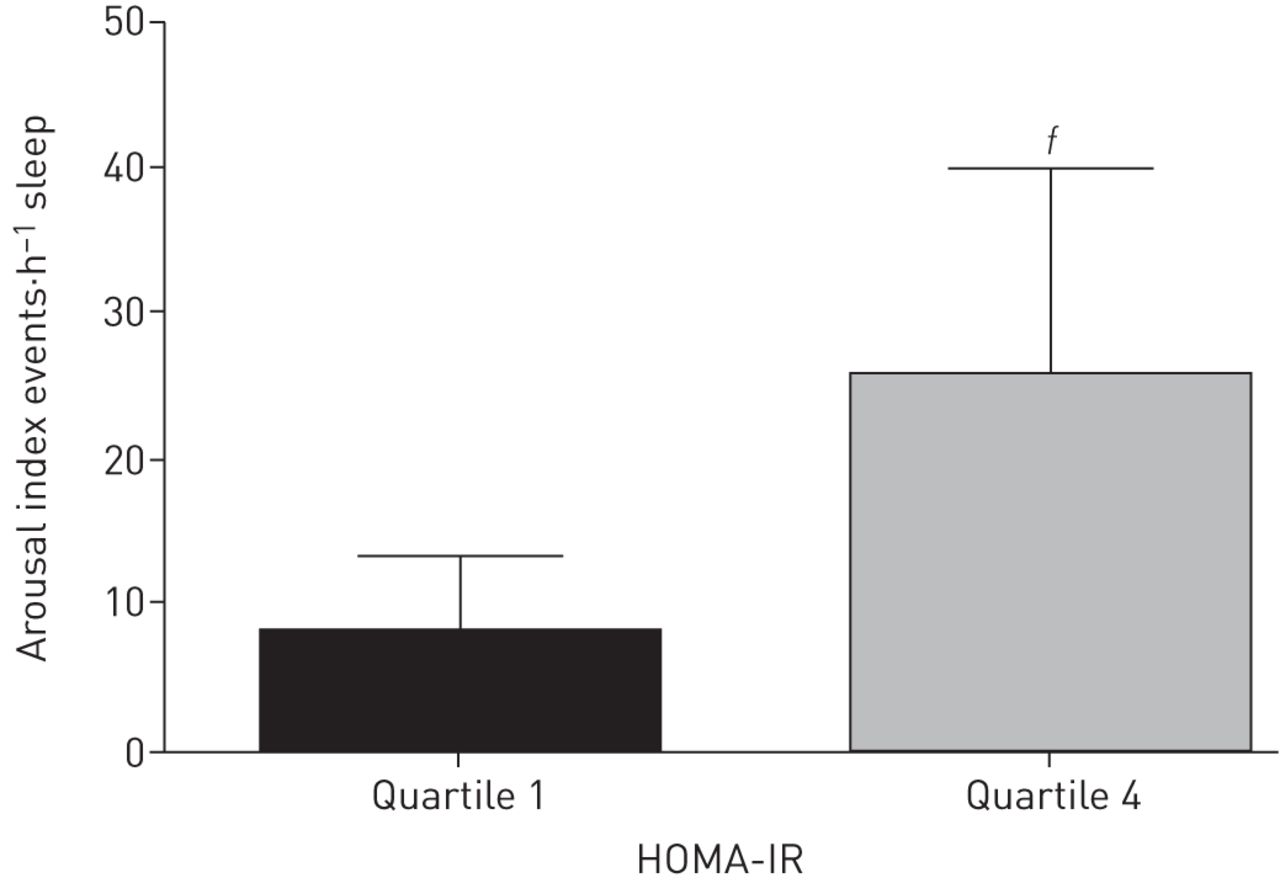
HOMA-IR quartile analyses are presented in supplementary table S3. Children in HOMA-IR quartile 4 (most insulin resistant) versus quartile 1 (least insulin resistant) had higher BMI and/or BMI z-score in all subgroups examined except nonobese children without OSA and obese children with moderate–severe OSA. In obese children with mild OSA, the AHI was significantly higher in quartile 4 versus quartile 1 (2.4±0.9 versus 1.7±0.9). In obese children with moderate–severe OSA, the respiratory ArI was near-significantly greater in those in quartile 4 versus quartile 1 (25.7±14.1 versus 8.1±5.2, p=0.053), clinically a very significant difference (figure 2).

{kind=link}
{kind=link}
Respiratory arousal index. Data represent mean±sd. Comparisons were made across the groups. ƒ: 0.1<p<0.05 for comparison between obese children with apnoea/hypopnoea index >5 events·h–1 in the lowest and highest homeostasis model assessment of insulin resistance (HOMA-IR) quartiles.
On multivariate linear regression analyses (table 6), spontaneous ArI significantly predicted FPI and HOMA-IR, albeit less than BMI. Total ArI was the strongest predictor of the McAuley index, with trends towards contributions from BMI and age. Finally, total sleep duration was the strongest contributor of FPG and HDL, with no contribution from either age or BMI. No other metabolic parameter was predicted by either OSA or sleep architecture measures after including BMI and/or age in regression modelling (data not shown).
Sleep variables predicting metabolic outcomes
Discussion
This study represents the largest cohort published to date examining relationships between OSA, sleep architecture and indicators of paediatric insulin resistance and the metabolic syndrome. We found that relationships between insulin resistance and dyslipidaemia were predominantly determined by adiposity rather than OSA or sleep architecture, but that sleep fragmentation contributed independently to insulin resistance and that total sleep duration was the primary, negative predictor of glucose and (to a limited extent) HDL, independently of obesity or OSA. Subcohort analyses further revealed that: 1) children with severe versus mild–moderate OSA were more obese, and among children with moderate–severe OSA, obese versus nonobese children had more severe sleep-disordered breathing; 2) OSA presence or degree did not impact insulin resistance, FPG, lipoprotein profiles or ratios among nonobese or obese children; 3) in obese children with mild OSA, higher AHI was seen in those with greater insulin resistance; and 4) sleep fragmentation due to respiratory arousals trended towards being higher in more insulin resistance obese children with moderate–severe OSA. Taken together, our findings suggest that obesity rather than OSA is the primary determinant of insulin resistance and metabolic dysfunction in young children, although OSA may further predispose to insulin resistance in obese children, that sleep fragmentation may contribute to insulin resistance independently of obesity and that insufficient sleep may predispose to higher glucose levels independently of either adiposity or OSA.
Impact of sleep duration, architecture and disturbance on glycaemia insulin resistance in nonobese and obese children
We found that obesity rather than OSA was the primary correlate of insulin resistance, but in obese children with mild OSA, higher AHI values were seen in the highest versus lowest HOMA-IR quartiles. These findings suggest a possible mild contributory effect of OSA to insulin resistance development in the context of obesity and its a priori predisposition to metabolic derangements. Similarly, and in agreement with our previous studies, significant associations emerged between TST and glucose independently of OSA or obesity, and between measures of insulin resistance and the proportion of total sleep time spent in REM and N3 sleep stages, a finding that has also been replicated in adults with OSA [23, 24].
Our findings that OSA is inconsistently associated with insulin resistance in children stand in contrast to a significant body of adult literature, wherein OSA associates with both insulin resistance [25] and T2DM [26, 27], and add to a body of contradictory paediatric literature: some studies show that OSA confers an increased risk of adverse metabolic states [9, 28], while we and others have found either no association with insulin resistance [29] or associations only in obese children [30].
Several explanations for the inconsistency of paediatric studies vis-à-vis results in adults present themselves. OSA and obesity in children may have existed for too short a time for metabolic comorbidities of either to predictably and consistently develop. Alternatively, the explanation for contradictory results in the paediatric literature may reside in puberty. Most of the paediatric studies showing a positive association of OSA with insulin resistance and the metabolic syndrome studied adolescents and, in the case of Kelly et al. [31], explicitly examined the contributory effect of puberty [9, 28, 29, 32]. The divergent findings in pre-pubertal children versus adolescents may be multifactorial: fat mass [33] and, in boys, intra-abdominal fat [34] increase significantly during puberty, predisposing to insulin resistance. Also, paediatric OSA appears to have two different phenotypic variants, sometimes termed OSA type I and OSA type II [35]. OSA type I, which predominates in younger children, relates primarily to lymphadenoidal hypertrophy, while OSA type II, which predominates in adolescents, primarily develops in the context of obesity. These phenotypes present similarly, but likely confer differing predispositions to metabolic sequelae [35]. Thus, associations between OSA and insulin resistance and the metabolic syndrome in adolescents versus younger children should be carefully delineated in future studies.
Sleep fragmentation and insulin resistance
Examination of the entire cohort showed that sleep fragmentation due to spontaneous arousals contributed to insulin resistance separately from obesity. Incidentally, we found that the McAuley index was a more sensitive measure of sleep fragmentation's impact upon insulin resistance than HOMA-IR. Our findings support a growing body of literature suggesting that sleep fragmentation may independently contribute to insulin resistance, both in the context of OSA [30, 36] and in healthy adults, where decreased insulin sensitivity and glucose disposal following experimental sleep fragmentation [37] suggest a causal link. Sleep fragmentation could impact insulin resistance through various potential mechanisms, including autonomic activation and increased cortisol levels [37]. Also, in murine models, we have found that experimental sleep fragmentation without hypoxaemia leads to visceral fat accumulation and insulin resistance [38, 39], accompanied by differentially expressed microRNA in a network of genes regulating insulin [40]. Sleep disruption should thus be minimised, especially in children at risk for insulin resistance. Future studies should be undertaken in children and adolescents with upper airway resistance syndrome (a condition of fragmented sleep without OSA) [41] to assess whether their insulin homeostasis is perturbed.
Strengths and limitations
Our study had a number of strengths. We had a very large cohort of children, allowing for subtle relationships to be explored, including the quartile analysis. Our study included a sizable number of both nonobese and obese subjects with and without OSA, allowing for determination of which relationships were dependent upon adiposity and which existed only in the presence of OSA. Additionally, OSA diagnosis and severity assessment was based upon gold standard in-laboratory full PSG.
Our study also had several limitations. All participants were referred for habitual snoring, introducing a possible referral bias. Also, we did not Tanner stage our participants and cannot rule out that some may have been pubertal. Furthermore, all metabolic measures examined reflected the fasting rather than post-prandial state. Additionally, as sleep duration in the sleep laboratory may not necessarily reflect home sleep duration, our findings of associations between sleep duration in the sleep laboratory and glycaemia are necessarily limited. While both HOMA-IR and the McAuley index have been validated against the gold standard insulin sensitivity measure from the hyperinsulinaemic–euglycaemic clamp technique, fasting insulin levels nonetheless reflect hepatic insulin sensitivity and gluconeogenesis; the association of OSA with skeletal muscle or adipose tissue insulin sensitivity and glucose disposal could not be assessed. 29 subjects had impaired fasting glucose, too few to analyse separately; separate associations could prevail in this subgroup. Also, we did not measure haemoglobin A1c, a measure of long-term glycaemic homeostasis, and cannot exclude the possibility that some subjects had hyperglycaemia not captured by fasting glucose. Finally, a number of subjects were excluded for low FPG or for FPI below assay sensitivity, forming an effective “floor” to the examination of associations between OSA and insulin resistance; more insulin-sensitive subjects may have been excluded. Finally, as this was not an intervention study, causality cannot be inferred.
Conclusion
In conclusion, we found that obesity rather than OSA seems to be the major driver of metabolic dysfunction in young children. OSA may have a small contribution, particularly in the context of obesity, and sleep fragmentation also appears to pose an additive risk for insulin resistance. We cannot rule out a small effect of OSA on metabolic function in nonobese children. Future studies are needed to better define the role of puberty in modulating OSA's adverse metabolic impact and the role of sleep fragmentation in induction of insulin resistance.
Acknowledgements
The authors would like to gratefully acknowledge Richa Kulkarni (Sleep Medicine Section, Dept of Pediatrics, Pritzker School of Medicine, Biological Sciences Division, The University of Chicago, Chicago, IL, USA) for her efforts in coordinating this study, the sleep technologists who performed the overnight polysomnograms, the laboratory technicians who drew the labs and performed the analyses, and finally the children and parents for their participation in this study.
Footnotes
For editorial comment see Eur Respir J 2016; 47: 1050–1053 [DOI: 10.1183/13993003.00115-2016].
This article has supplementary material available from erj.ersjournals.com
Support statement: This study was supported by National Institutes of Health grant HL-65270. Funding information for this article has been deposited with FundRef.
Conflict of interest: None declared.
- Received August 27, 2015.
- Accepted November 30, 2015.
- Copyright ©ERS 2016
References










