





Abstract
Activin-A is a pleiotropic cytokine that regulates allergic inflammation. Its role in the regulation of angiogenesis, a key feature of airways remodelling in asthma, remains unexplored. Our objective was to investigate the expression of activin-A in asthma and its effects on angiogenesis in vitro.
Expression of soluble/immunoreactive activin-A and its receptors was measured in serum, bronchoalveolar lavage fluid (BALF) and endobronchial biopsies from 16 healthy controls, 19 patients with mild/moderate asthma and 22 severely asthmatic patients. In vitro effects of activin-A on baseline and vascular endothelial growth factor (VEGF)-induced human endothelial cell angiogenesis, signalling and cytokine release were compared with BALF concentrations of these cytokines in vivo.
Activin-A expression was significantly elevated in serum, BALF and bronchial tissue of the asthmatics, while expression of its protein receptors was reduced. In vitro, activin-A suppressed VEGF-induced endothelial cell proliferation and angiogenesis, inducing autocrine production of anti-angiogenic soluble VEGF receptor (R)1 and interleukin (IL)-18, while reducing production of pro-angiogenic VEGFR2 and IL-17. In parallel, BALF concentrations of soluble VEGFR1 and IL-18 were significantly reduced in severe asthmatics in vivo and inversely correlated with angiogenesis.
Activin-A is overexpressed and has anti-angiogenic effects in vitro that are not propagated in vivo, where reduced basal expression of its receptors is observed particularly in severe asthma.
Abstract
Anti-angiogenic role of activin-A, overexpressed in severe asthma, may be compromised by reduced receptor signalling http://ow.ly/W4kxF
Introduction
Severe asthma is increasingly recognised as a distinct disease entity associated with high morbidity and healthcare costs, and represents a major unmet therapeutic need [1]. In addition to airways inflammation, remodelling changes are implicated in the pathogenesis of severe asthma and can lead to irreversible airflow obstruction [2]. Local airways angiogenesis is increasingly acknowledged to play a role in the pathophysiology of airway remodelling changes associated with asthma [3]. Disordered angiogenesis may occur early in life and lead to accelerated lung function decline, prolonged symptoms, impaired quality of life and even death [3, 4]. Thus, amelioration of this process represents a probable valid target for therapeutic intervention, and is indeed already the subject of elaborate research [5, 6].
Angiogenesis is a multifaceted process regulated by the balance between the actions of numerous pro- and anti-angiogenic factors [7–12] such that no single mediator can be considered solely responsible [13]. Vascular endothelial growth factor (VEGF) promotes endothelial cell proliferation and differentiation and represents a key inducer of bronchial angiogenesis in asthma through the activation of its membrane receptors, VEGFR1 and VEGFR2 [14, 15]. Other pro-angiogenic factors, such as interleukin (IL)-17 and IL-13, have also been identified [10, 16, 17]. Among angiogenesis inhibitors, soluble VEGF receptor (sVEGFR)1, IL-18 and IL-32 exhibit potent potential effects, although their possible roles in the context of severe asthma have not been explored [7–9, 18, 19]. Understanding the balance of interactions between pro- and anti-angiogenic activities in the inflammatory milieu of the severe asthmatic airways remains a considerable challenge.
Activin-A is a pleiotropic cytokine, belonging to the transforming growth factor (TGF)-β superfamily [20]. While TGF-β1 signalling occurs via the type II TGFβ receptor, activin-A signals primarily through binding to the activin-A receptor type IIA (Act-RIIA), which results in phosphorylation of the type I receptor (termed activin-like kinase (ALK)-4) [20]. Downstream signalling occurs via phosphorylation of receptor-regulated Smads that translocate to the nucleus to initiate gene transcription [20]. Both TGF-β and activin-A signalling pathways have been shown to be activated upon allergen provocation in asthma [21, 22]. Emerging evidence underscores the potential impact of activin-A on the regulation of airway inflammation and remodelling [22–27]. Previous studies by our group and others have demonstrated an anti-inflammatory role for activin-A in various settings [28–31], while other studies have shown opposing effects [23, 24]. Interestingly, although the effects of activin-A on airway vascularisation have never been addressed, investigation of its angiogenic effects on other tissues has also produced conflicting results [32–34]. These discrepancies highlight the possibility that although activin-A may be overexpressed in inflammation, its end-effects may be diverse or even ablated by modification of its signalling in particular inflammatory environments in vivo.
In the present study, we set out to investigate the expression and possible role of activin-A in regulating airways vascularity in severe asthma. We hypothesised that activin-A is produced to excess in the asthmatic bronchial mucosa by a variety of infiltrating and structural cells, including vascular endothelial cells. We further hypothesised that activin-A has the potential to regulate endothelial cell proliferation and angiogenesis but that its end-effects are influenced by the integrity of its signalling pathway and the presence of other locally expressed mediators. To achieve this, we investigated the expression and origins of activin-A and its signalling molecules in the airways of patients with asthma of varying severity as well as healthy controls. We also examined both the production of activin-A and its interactions with other cytokines regulating angiogenesis in cultured human endothelial cells in vitro.
Methods
Full methodological details are provided in the online supplementary material.
Study population
57 nonsmoking subjects, including 16 nonatopic healthy controls, 19 patients with persistent mild-to-moderate asthma (MMA) and 22 patients with persistent severe asthma (SA) participated in the study (table 1). All asthmatic patients had a physician-confirmed diagnosis and asthma severity was defined according to recent Global Initiative for Asthma and European Respiratory Society (ERS)/American Thoracic Society guidelines [35, 36]. Patients had been taking anti-asthmatic therapy at a stable dosage for ≥3 months prior to study enrolment. Subjects underwent fibre-optic bronchoscopy with endobronchial biopsy and bronchoalveolar lavage fluid (BALF) collection at steady state. Serum samples were collected from patients at steady state and if and when an exacerbation occurred during a 2-year follow-up period (n=17), prior to the administration of oral corticosteroids (online supplementary fig. E1). Asthma exacerbations were defined as episodes of severe symptom deterioration and elevated rescue medication usage that required the administration of systemic corticosteroids or an elevated dosage for ≥3 days [37]. Prior to participation in the study, all subjects provided informed written consent and the protocol was approved by the hospital's research ethics committee and the Greek National Organization for Medicines. All studies were conducted according to the principles of the Declaration of Helsinki. More information regarding the study population is available in the online supplementary material.
Subject characteristics and results of quantification of activin-A, activin-A receptor type IIA (Act-RIIA), activin-like kinase (ALK)-4 and airway angiogenesis in all groups examined
Fibre-optic bronchoscopy and sample collection
Bronchoscopy was performed on an outpatient basis, as previously described [38]. After inspection of the bronchial tree, bronchoalveolar lavage was performed and samples with a fluid recovery of ≥60% were retained for further analysis (healthy controls n=13, MMA n=12 and SA n=19), according to the ERS task force guidelines regarding measurements of acellular components in BAL [39].
Cytokine measurements
Commercially available ELISA kits were used to determine the concentration of human activin-A, IL-17A, IL-18, IL-32, VEGF and sVEGFR1 in the serum, BALF and endothelial cell culture supernatants according to the manufacturers' instructions (online supplementary material).
Tissue staining and measurements
Paraffin sections 3–4 μm thick were used for haematoxylin and eosin and immunohistochemical staining and 6-μm-thick frozen sections were used for immunofluorescence. Immunostaining for activin-A, ALK-4 and Act-RIIA, and subsequent quantification were performed as previously described [22, 30, 38]. Immunofluorescent staining for cellular markers was also performed as preciously described [38]. Blood vessels were identified using the vascular endothelial cell marker CD31 (goat polyclonal anti-CD31, sc-1506, 1:200; Santa Cruz, Dallas, TX, USA), and the total number of vessels was divided by the biopsy area to determine the numbers of vessels per unit area (mm2) of the sections. Extensive methodological information on tissue processing and analysis is presented in the online supplementary material.
Endothelial cell cultures, proliferation and in vitro angiogenesis assays
Human umbilical vein (HUVEC) and human pulmonary microvascular (HPMEC) endothelial cell lines were purchased from Merck Millipore (Darmstadt, Germany) and PromoCell (Heidelberg, Germany), respectively. Their proliferation was measured 48 h after plating (>90% confluence) using a commercially available kit (WST-8; Cayman Chemical, Ann Arbor, MI, USA), as previously described [40]. For the in vitro angiogenesis assay, the V2A kit (ZHA 4000; Cellworks, Buckingham, UK) was employed as previously described [40]. Recombinant human (rh)-IL-13 (10 ng·mL−1; R&D Systems, Wiesbaden, Germany), rh-activin-A (200 ng·mL−1; R&D Systems) and rh-VEGF (2 ng·mL−1; Biolegend, San Diego, CA, USA) were applied for 48 h at concentrations similar to previously described studies [34, 41] after performing dose–response and kinetic studies (online supplementary fig. E2). A human polyclonal anti-activin-A antibody was used for blocking activin-A (15 μg·mL−1; R&D Systems). All experiments were repeated three times in tetraplicate wells. The results were expressed as percentages of control medium values.
RNA extraction and real-time PCR
Total RNA was isolated from HUVEC in TRI Reagent (MRC, Cincinnati, OH, USA) following the manufacturer's instructions (online supplementary table E1).
Statistical analysis
Data are expressed as median and interquartile range, unless specified otherwise. Comparisons between subject groups were performed using the Kruskal–Wallis one-way ANOVA accompanied by Dunn's post hoc correction. The Wilcoxon matched-pair test was applied to compare activin-A expression in the stable state and during exacerbation. Correlation coefficients were calculated using the Spearman's rank method. Regarding the in vitro data, comparisons between three or more groups were analysed by one-way ANOVA followed by the Mann–Whitney U-test to compare specific pairs for significant differences. A statistical software package was used for all data analysis and graph preparation (Prism v5; GraphPad, San Diego, CA, USA). A p-value of ≤0.05 was considered significant.
Results
For median values and interquartile ranges please refer to table 1.
Activin-A is increased in the airway of asthmatics and peaks during asthma exacerbations
Activin-A concentrations were measured in the serum and BALF of asthmatics with distinct disease severities. Patients with MMA and severe asthma had significantly elevated serum activin-A compared to healthy controls (fig. 1a). In addition, activin-A was significantly increased in the BALF of patients with MMA and severe asthma compared to healthy controls (fig. 1b). Interestingly, activin-A was markedly further increased in the serum of asthmatics during exacerbations compared with the steady-state (fig. 1c). No associations were found between activin-A levels in the serum, BALF or bronchial tissue and the risk of exhibiting an exacerbation among asthma patients (data not shown). Still, there was a trend for a negative association between body mass index and activin-A levels in the serum and BALF; however this did not reach statistical significance (online supplementary fig. E3).

Activin-A is increased in asthma. Activin-A concentrations in a) serum and b) bronchoalveolar lavage fluid (BALF) at steady state, and c) serum during exacerbation. d) Representative microphotographs demonstrating activin-A expression: i) isotype control; ii) healthy control; iii) severe asthmatic; iv) severe asthmatic. Scale bars=20 μm. Blue arrow: epithelial staining; red arrows: subepithelial staining. e) and f) Quantification of activin-A+ cells in the bronchial mucosa. g–j) Confocal microscopic images from bronchial tissue of severe asthmatics demonstrating colocalisation (yellow) of activin-A (red) expression by subepithelial cell markers (green); specifically g) mast cells, h) neutrophils, i) macrophages and j) smooth muscle cells. Scale bars=5 μm. Data are presented as median (interquartile range). HC: healthy controls; MMA: mild/moderate asthmatics; SA: severe asthmatics; DAPI: 4′,6-diamidino-2-phenylindole; SMA: smooth muscle actin. *: p<0.05; **: p<0.01; ***: p<0.001.
Expression of activin-A immunoreactivity was markedly elevated in epithelial and subepithelial infiltrating inflammatory cells in asthmatics (fig. 1d). Quantitative analysis revealed that the median number of activin-A immunoreactive cells was significantly increased in the bronchial epithelium in patients with MMA and severe asthma, compared to healthy controls (fig. 1e). Furthermore, activin-A expression by resident and infiltrating cells in the lamina propria was significantly elevated in severely asthmatic subjects compared to both MMA and healthy controls (fig. 1f). No significant differences were observed in the serum, BALF or airway expression of activin-A between atopics and nonatopic asthmatics (online supplementary fig. E4) or in the severe asthmatics treated or not treated with oral corticosteroids (online supplementary fig. E5). Double immunofluorescence coupled with confocal microscopy in severe asthmatics revealed that mast cells, neutrophils, macrophages and endothelial and smooth muscle cells express activin-A, which in some cases seems to have intranuclear localisation (fig. 1g–j).
Activin-A signalling pathways are dysregulated in severe asthma
In contrast to activin-A, bronchial mucosal expression of immunoreactivity for its receptor proteins was reduced, especially in severe asthmatics. The median number of cells expressing Act-RIIA immunoreactivity was significantly reduced in the bronchial epithelium and lamina propria in MMA and severely asthmatic subjects compared to healthy controls (fig. 2a). ALK-4 expression was significantly reduced in epithelial and subepithelial cells only in severe asthma compared to healthy controls (fig. 2b), with intermediate expression in MMA. No significant differences were observed in the airway expression of activin-A and its receptors ALK-4 and Act-RIIA between atopic and nonatopic asthmatics (online supplementary fig. E4) nor in the severe asthmatics treated or not treated with oral corticosteroids (online supplementary fig. E5).

Expression of activin-A receptors activin-A receptor type IIA (Act-RIIA) and activin-like kinase (ALK)-4 is markedly reduced in severe asthmatics. Representative microphotographs and quantitative analysis of a) Act-RIIA and b) ALK-4 in bronchial biopsy sections. i) Healthy controls; ii) mild/moderate asthmatics; iii) severe asthmatics; iv) expression in epithelial cells; v) subepithelial cells. Data are presented as median (interquartile range). HC: healthy controls; MMA: mild/moderate asthmatics; SA: severe asthmatics. Scale bars=20 μm. *: p<0.05; **: p<0.01; ***: p<0.001.
Activin-A in the asthmatic bronchial mucosa correlates with angiogenesis
Evaluation of the vascularity of the bronchial mucosa demonstrated that MMA and severely asthmatic patients had significantly elevated median numbers of blood vessels per unit area in the lamina propria compared to healthy controls (fig. 3a). Interestingly, our data revealed a strong correlation between the degree of angiogenesis and activin-A expression in epithelial (fig. 3b) and subepithelial cells (fig. 3c). In contrast, bronchial mucosal expression of both activin-A receptors, ALK-4 and Act-RIIA, inversely correlated with angiogenesis (fig. 3d–g), suggesting that activin-A signalling is decreased in patients with prominent angiogenesis. Airways angiogenesis inversely correlated with forced expiratory volume in 1 s, and although no correlation was observed with activin-A, increased airways expression of ALK-4 and Act-RIIA was associated with improved lung function (online supplementary fig. E6).

Airway angiogenesis correlates with activin-A expression and is associated with reduced tissue expression of activin-like kinase (ALK)-4 and activin-A receptor type IIA (Act-RIIA). a) Quantification of airway angiogenesis; correlation of angiogenesis with b) epithelial and c) subepithelial activin-A expression; inverse correlations of angiogenesis with d) epithelial and e) subepithelial expression of ALK-4; and f) epithelial and g) subepithelial expression of Act-RIIA. Data are presented as median (interquartile range). HC: healthy controls; MMA: mild/moderate asthmatics; SA: severe asthmatics. **: p<0.01; ***: p<0.001.
Activin-A suppresses spontaneous and VEGF-induced proliferation of human pulmonary endothelial cells
Immunofluorescence analysis of biopsies from severe asthmatics revealed that the majority of mucosal peribronchial endothelial cells expressed immunoreactivity for activin-A (fig. 4a). ALK-4 and Act-RIIA were also expressed, albeit by fewer of the pulmonary epithelial cells (fig. 4b and data not shown).

Activin-A (Act-A) inhibits the proliferation of human pulmonary microvascular endothelial cells (HPMEC) in vitro. Representative photomicrographs demonstrating a) Act-A (red) and b) activin-like kinase (ALK)-4 (red) in CD34+ epithelial cells (green) in severe asthmatics. Colocalisation is shown in yellow (arrows). Scale bars=10 μm. c) Representative photomicrographs for i) isotype control; ii) von Willebrand factor (vWF); iii) Act-A; iv) ALK-4; v) activin-A receptor type IIA (Act-RIIA); and vi) phospho (p)Smad2/3 on HPMEC in the presence or absence of recombinant human (rh)-interleukin (IL)-13. Scale bars=20 μm. d) Quantification of immunoreactivity on HPMEC in the presence (+) or absence (-) of rh-IL-13. e) Act-A concentrations in HPMEC supernatants in the presence or absence of IL-13. f) Proliferation of HPMEC. Data are presented as mean±sem from three independent experiments in tetraplicate wells. DAPI: 4′,6-diamidino-2-phenylindole; rAct-A: recombinant activin-A; aAct-A: anti-activin-A; VEGF: vascular endothelial growth factor. *: p<0.05; **: p<0.01; ***: p<0.001.
Corresponding in vitro experiments using HUVEC and, more pertinently, HPMEC [42] confirmed that, as in vivo, immunoreactivity for activin-A and its receptors was expressed in the resting state by subpopulations of HPMEC (fig. 4c and d) and HUVEC (not shown). To mimic the pro-allergic in vivo setting, we exposed HPMEC to IL-13, a critical inflammatory mediator in asthma with an active signalling pathway in endothelial cells [43, 44]. IL-13 exposure significantly increased the mean percentages of HPMEC expressing activin-A, but decreased the mean percentages of ALK-4+ and Act-RIIA+ cells (fig. 4d). Moreover, activin-A was detectable in culture supernatants of HPMEC at rest and its secretion significantly enhanced by IL-13 (fig. 4e). Exposure of endothelial cells to exogenous activin-A significantly reduced both spontaneous and VEGF-enhanced HPMEC proliferation, even in the presence of the inflammatory cytokine, IL-13 (fig. 4f). Similar findings were obtained using HUVEC (data not shown). In an opposite approach, we evaluated the effects of activin-A neutralisation on HPMEC proliferative responses. Blockade of activin-A resulted in enhanced HPMEC responses to VEGF and IL-13 (fig. 4f).
Activin-A inhibits VEGF-induced angiogenesis
To further explore the effects of activin-A on the regulation of human endothelial cell responses, we utilised an in vitro model of angiogenesis. In this assay, exogenous VEGF strongly promoted spontaneous angiogenesis, reflected by a significant elevation of all output parameters investigated, such as the number of junctions and tubules, as well as the total tubule length (fig. 5a and b). Activin-A did not alter spontaneous angiogenesis but significantly inhibited VEGF-induced angiogenesis (fig. 5b). Interestingly, activin-A significantly reduced VEGF-enhanced angiogenesis in the presence of IL-13 (fig. 5b), while neutralisation of activin-A resulted in enhanced angiogenic responses to VEGF and IL-13 (fig. 5c).

Activin-A (Act-A) inhibits vascular endothelial growth factor (VEGF)-induced angiogenesis in vitro. a) Representative photomicrographs showing the formation of endothelial tubular structures (black) at day 14, as indicated. Scale bars=500 μm. Computer-assisted quantification of the number of junctions, tubules and tubule length demonstrating the effects of stimulation with b) anti-(a)Act-A neutralising antibody or Ig control and c) recombinant (r)Act-A on VEGF- and interleukin (IL)-13-induced angiogenic processes. Data are presented as mean±sem from six different photomicrographs per parameter and are representative of three independent experiments in tetraplicate wells. *: p<0.05; **: p<0.01; ***: p<0.001.
Activin-A regulates the balance of VEGF receptors and induces an anti-angiogenic cytokine milieu
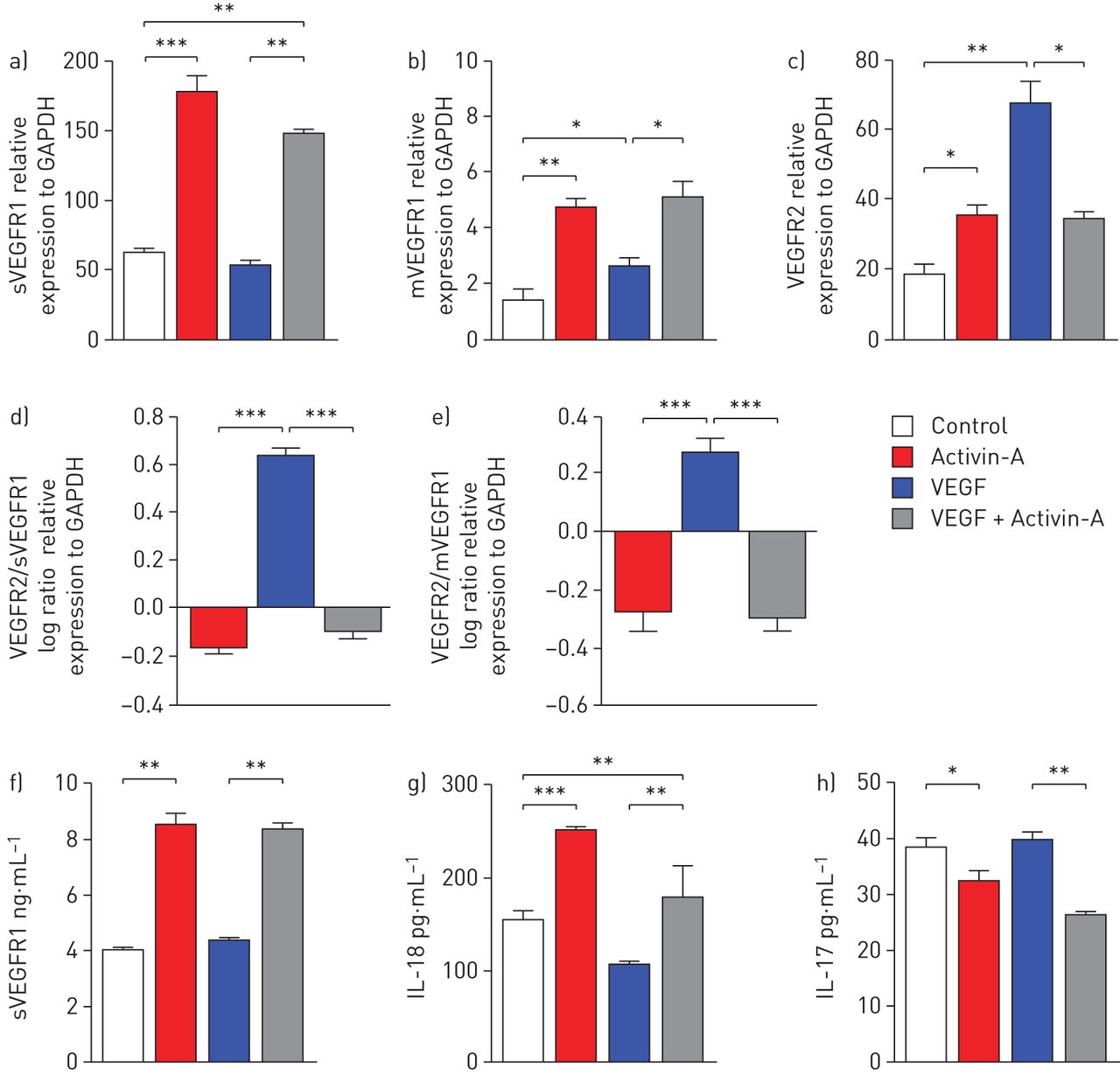
Stimulation of HUVEC with activin-A resulted in a significant (approximately three-fold) increase in expression of mRNA encoding sVEGFR1 and membrane-bound (m)VEGFR1, and to lesser extent VEGFR2, compared with controls (fig. 6a and b). VEGF significantly increased expression of mRNA encoding VEGFR2, while it had no discernible effects on mRNA encoding sVEGFR1 (fig. 6a and c). When employed together, the effects of activin-A prevailed over those of VEGF (fig. 6a–c); where VEGF induced a high VEGFR2/sVEGFR1 mRNA ratio, this was reversed in the presence of activin-A (fig. 6d). Similar findings were observed with the VEGFR2/mVGEFR1 ratio (fig. 6e). Consistent with the mRNA data, stimulation of endothelial cells with activin-A significantly increased the release of sVEFGR1 protein into the culture supernatants in the presence or absence of VEGF (fig. 6f). No significant differences were observed in VEGFR2 protein levels between the groups studied (data not shown).

The anti-angiogenic effects of activin-A are associated with enhanced soluble (s) vascular endothelial growth factor receptor (VEGFR)1 and decreased VEGFR2. Quantitative real-time PCR analysis of a) sVEGFR1; b) membrane-bound (m)VEGFR1; and c) VEGFR2 mRNA expression by human umbilical vein epithelial cells (HUVEC) relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) upon stimulation for 96 h. d) Log ratios, normalised to control, of VEGFR2/sVEGFR1 and e) VEGFR2/mVEGFR1 mRNA expression, relative to GAPDH. f) sVEGFR1 concentrations in culture supernatants. g) Interleukin (IL)-18 and h) IL-17 concentrations in culture supernatants. Data are presented as mean±sem from three independent experiments in duplicate wells. *: p<0.05; **: p<0.01; ***: p<0.001.
Incubation of endothelial cells with activin-A in vitro resulted in significantly elevated mean release of IL-18 into culture supernatants, compared with control medium, whereas VEGF significantly reduced mean IL-18 release (fig. 6g). In the additional presence of activin-A this effect of VEGF was reversed (fig. 6g). In contrast, activin-A significantly reduced mean release of the pro-angiogenic cytokine IL-17 in control medium and VEGF-treated cells (fig. 6h). IL-32 release by human epithelial cells was not altered in the presence of activin-A or VEGF (online supplementary fig. E7).
IL-18 and sVEGFR1 are decreased in the BALF and inversely correlate with bronchial tissue angiogenesis in severe asthma
In an effort to translate our findings into the clinical setting, we investigated the concentrations of the above-mentioned mediators in the BALF of patients with asthma of varying severity. The median concentration of IL-18 and sVEGFR1 were significantly lower in the BALF of severely asthmatic patients compared to healthy controls (fig. 7a and b). Conversely, VEGF was significantly increased in severe asthma (fig. 7c). No significant differences were observed in BALF median concentrations of IL-17 and IL-32 between the groups examined (online supplementary fig. E8). In contrast to VEGF, BALF concentrations of IL-18 and sVEGFR1 inversely correlated with the degree of airway angiogenesis (fig. 7d–f). Moreover, IL-18 and sVEGFR1 levels in the BALF tended to inversely correlate with those of activin-A (online supplementary fig. E9). No significant correlations were found between VEGF, IL-18 and sVEGFR1 and activin-A levels in the serum (data not shown).

Bronchoalveolar lavage fluid (BALF) interleukin (IL)-18 and soluble (s) vascular endothelial growth factor receptor (VEGFR)1 concentrations inversely correlate with airways vascularity in severe asthmatics (SA). a) sIL-18; b) sVEGFR1; and c) VEGF concentrations in the BALF. Correlations of d) IL-18; e) sVEGFR1; and f) VEGF with angiogenesis in the airways of all subjects. Data are presented as median (interquartile range). HC: healthy controls; MMA: mild/moderate asthmatics. *: p<0.05; **: p<0.01.
Discussion
Our study clearly demonstrates that activin-A is increased in the serum, BALF and bronchial tissue in patients with asthma and further increases during exacerbations. Our preliminary data further suggest that its mass production is not susceptible to systemic glucocorticoid inhibition. The expression of activin-A is more prominent in the subepithelium of the bronchial mucosa of severe asthmatics, where it is produced by a variety of structural and inflammatory cells, including bronchial endothelial cells in a “T-helper type II-high” environment. The hypothesis that endothelial cells are a major source of activin-A production in asthma in vivo is supported by our observed correlation between airways vascularity (the degree of which was, in turn, associated with more severe disease) and activin-A expression. Bronchial epithelial cells also appear to be a potent source. Our hypothesis that activin-A may exert regulatory effects on angiogenesis in asthma in vivo is supported by the fact that endothelial cells express activin-A receptors, albeit in a potentially attenuated form in the more severe patients.
No significant differences regarding activin-A levels were observed in the serum or BALF of MMA and severely asthmatic patients. That the expression of certain mediators involved in severe asthma and associated airway remodelling processes does not differ between severe and mild/moderate asthmatics is not uncommon, as in the case of matrix metalloproteinases, eotaxin, IL-5 and IL-13 [45–47], or even in the case of established biomarkers, such as sputum eosinophils and exhaled nitric oxide fraction [46, 48]. Nevertheless, as in the case of IL-13 [47], our findings did reveal a significant increase in the numbers of resident and infiltrating cells expressing activin-A in the lamina propria of severe asthmatics. The observation that activin-A exerts effects on endothelial cells in the airways, even though it is predominantly expressed by bronchial epithelial cells, corroborates the pleiotropic nature of activin-A in the context of asthma. Predominance of expression of a certain cytokine by the bronchial epithelium does not preclude a pathogenic role for the corresponding factor in other cell types and processes. In fact, several mediators involved in asthma pathophysiology, such as TGF-β [49, 50], osteopontin [38, 51], pentraxin-3 [52] and resistin-like molecule-β [53] are prominently expressed in the bronchial epithelium and exert effects on subepithelial cells. Interestingly, and in agreement with other studies, our findings demonstrated that activin-A is also localised in the nucleus of subepithelial cells in the bronchial mucosa, possibly reflecting other, unidentified roles for this cytokine in gene regulation [54, 55].
A prominent finding of our study is that activin-A has the ability to suppress human pulmonary endothelial cell proliferation and angiogenesis in vitro, even in the presence of a robust endothelial cellular activator, such as VEGF. VEGF exerts its angiogenic effects in the asthmatic airway through its two main receptors VEGFR1 and VEGFR2 [14], which have similar structures but divergent functions in vivo [18]. We demonstrate here that the anti-angiogenic effects of activin-A may be propagated partly through its ability to invert the ratio of VEGFR2/VGEFR1 mRNA expression by endothelial cells, thereby modulating VEGF signalling. Furthermore, we show that activin-A stimulation has the potential to alter endothelial cell production of cytokines that affect angiogenesis, in particular by increasing the secretion of the anti-angiogenic IL-18 [7, 9] while reducing that of the pro-angiogenic IL-17 [10], both at baseline and in the presence of VEGF. Finally, we also demonstrate an additional novel biological effect of activin-A on the production of the inhibitory circulating form of the VEGR1 by endothelial cells, through which it may indirectly further mitigate VEGF signalling [19].
The impact of activin-A on bronchial angiogenesis has not previously been explored, although its effects on vascularisation have been investigated in other tissues with conflicting results [32–34]. Our data throw considerable light on the possible reasons for these conflicting data. The fact that angiogenesis appears to proceed in severe asthmatics in the face of elevated expression of activin-A raises the possibility that its effects may not be propagated at least partly because of modification of its signalling pathway. This hypothesis is supported by our finding that in contrast to activin-A, the expression of its receptors, ALK-4 and Act-RIIA was reduced, at least at the level of reduced immunoreactivity, in the bronchial tissue of severe asthmatics.
It is possible that these changes are brought about by the highly inflammatory environment of the bronchial mucosa in asthma. In support of this, we show that stimulation with IL-13 markedly reduces the expression of Act-RIIA and, to a lesser extent, ALK-4 in human pulmonary endothelial cells. IL-13 is a critical inflammatory mediator in asthma with an active signalling pathway in endothelial cells [43, 44] and the ability to induce VEGF production [16, 17] and mediate its effects in vivo [56]. Similarly, inhibition of activin-A signalling by other inflammatory mediators locally in the airways, such as lipopolysaccharide and tumour necrosis factor-α has been reported previously [57]. A variety of cytokines with increased expression in the bronchial mucosa of severe asthmatics, including VEGF as observed herein and by others [11], may counteract the upregulation of angiogenic inhibitors, such as sVEGFR1 and IL-18, through which activin-A partly mediates its effects. Our observation that both sVEGFR1 and IL-18 concentrations were decreased in the BALF of severe asthmatics, whereas that of VEGF was increased even in the presence of high concentrations of activin-A, are in agreement with this hypothesis (fig. 8). Decreased ALK-4 expression has been also reported in the airway epithelium of chronically challenged mice [58], suggesting that allergen exposure per se could modify the expression pattern of activin-A's signalling pathway. Although we observed no differences regarding the expression of activin-A, ALK4 and Act-RIIA in the lung biopsies from atopic and nonatopic asthmatics, the possibility that allergen exposure influences their expression cannot be excluded.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
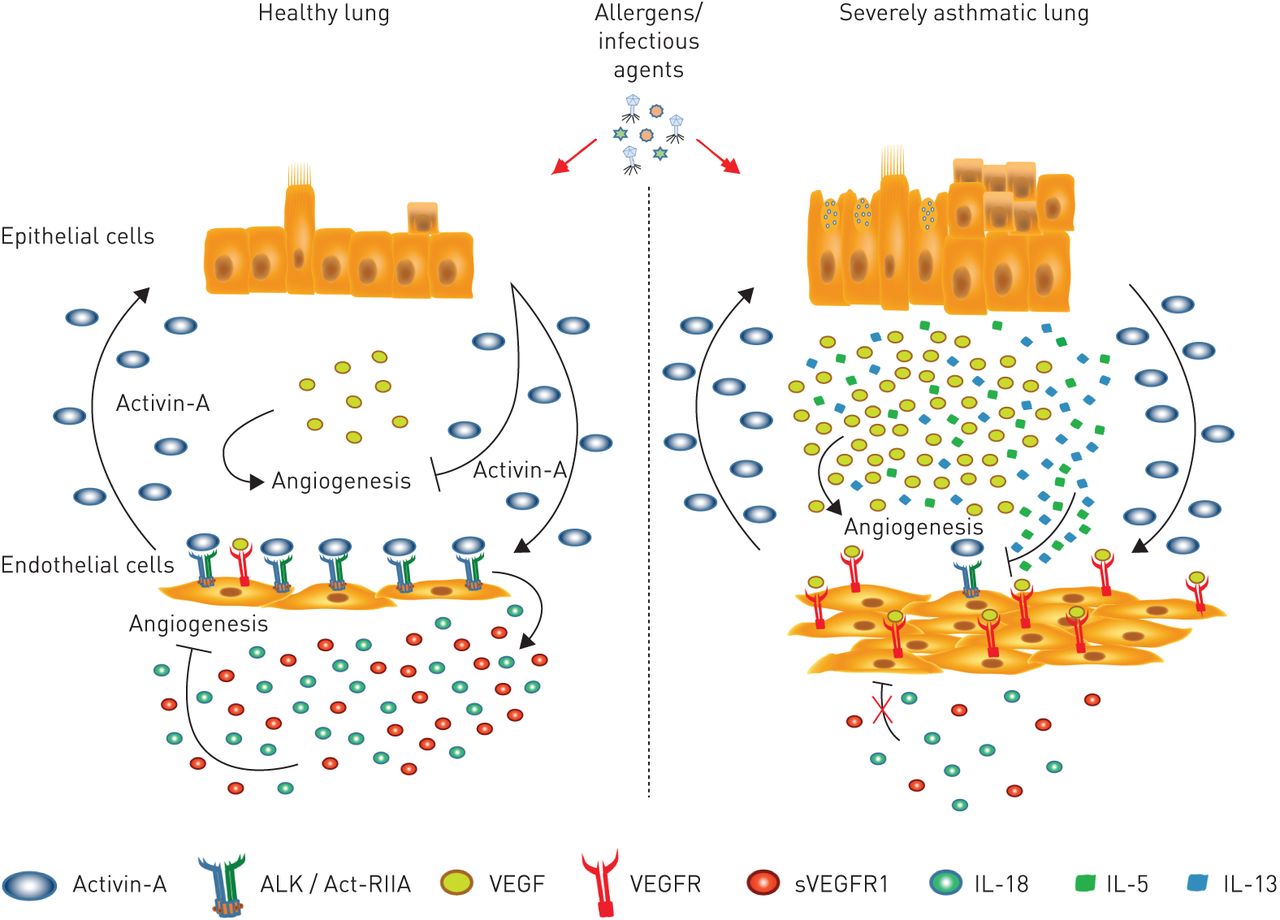
Proposed model for activin-A-mediated inhibition of angiogenesis in the airways of asthmatic individuals. In healthy individuals, upon exposure to environmental antigens, bronchial epithelial cells release activin-A, which signals through its receptors on airway endothelial cells and inhibits vascular endothelial growth factor (VEGF)-induced angiogenesis. This is associated with an enhanced release of the anti-angiogenic mediators interleukin (IL)-18 and VEGF receptor (R)1. However, in asthmatics, in the presence of inflammatory cytokines in the airways (such as IL-13, IL-5, etc.) the expression of activin-A receptors is decreased and, thus, its signalling pathways are disrupted. As a result, activin-A, although overexpressed in the airways of asthmatics, cannot inhibit VEGF-induced angiogenesis or increase IL-18 and VEGFR1 levels. ALK: activin-like kinase; Act-RIIA: activin-A receptor type IIA; s: soluble.
The current study presents certain limitations. In an observational cross-sectional study, there is no longitudinal follow-up and it is difficult to pursue specific postulated mechanisms of disease, for which longer-term studies and/or “models” may sometimes be more appropriate. Secondly, the activin-A concentrations used in our study for the stimulation of endothelial cell lines, although optimised by preliminary in vitro experiments and similar to those used by others, are higher than those observed in the BALF and serum of our asthma patients ex vivo. Finally, we did not use fresh primary bronchial endothelial cells for our in vitro experiments and, in the absence of an established cell line based on human peribronchial endothelial cells, we have used a human pulmonary microvascular endothelial cell line, as the most appropriate readily available cell line for studying human lung diseases [42, 59, 60].
In summary, our studies expand our knowledge of the scope of activities of activin-A in asthma, adding potential anti-angiogenic effects to its already versatile functions as a cytokine. We also demonstrate the principle, probably applicable to any complex chronic inflammatory process, that overexpression of a cytokine such as activin-A in asthma does not inevitably result in the same end-effects as observed in vitro because of complex, local environmental interactions in vivo. Our findings provide the groundwork for future research aiming to better understand these complex interactions in order to evaluate activin-A as a possible target for affecting the course of angiogenesis in asthma.
Acknowledgements
The authors are grateful to all subjects who participated in this study. The authors would like to thank Anna Agapaki, Eleni Rigana and Stamatis Pagakis (Histochemistry Unit and Biological Imaging Facility, Biomedical Research Foundation of the Academy of Athens, Athens, Greece) for their expert technical assistance with confocal imaging and Cailong Fang (King's College London School of Medicine, London, UK) for his valuable guidance regarding the in vitro experiments. The authors would also like to express their gratitude to Peprotech EC Ltd (London, UK) and especially to Anna Seymour and Almira Bartolome for their generosity during a time of financial distress.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: K. Samitas was supported by a research grant from the Hellenic Thoracic Society. G. Xanthou was supported by the Hellenic Ministries of Health and Education and by the “Romain Pauwels Excellence Award” from the European Respiratory Society. S. Tousa was supported by a Fondation Santé biomedical research grant. N. Poulos was supported by an European Federation of Immunological Societies fellowship.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received March 17, 2015.
- Accepted December 4, 2015.
- Copyright ©ERS 2016
References










