





Abstract
PTTM is a rare, likely under-diagnosed cause of PH, which is related to, but distinct from PTE http://ow.ly/OQad5
To the Editor:
Pulmonary hypertension (PH) is defined as a mean pulmonary artery pressure ≥25 mmHg, and can be associated with multiple conditions. The diagnostic strategy and treatment of PH is heavily reliant on accurately classifying patients [1]. We recently identified two patients presenting with rapidly fatal PH of unknown aetiology, who were subsequently diagnosed at post mortem with pulmonary tumour thrombotic microangiopathy (PTTM). This is a rare, albeit likely under-diagnosed cause of PH, characterised by a remodelling pulmonary vasculopathy rather than simple tumoural obstruction. At present, it is unclear where PTTM fits within the current classification system.
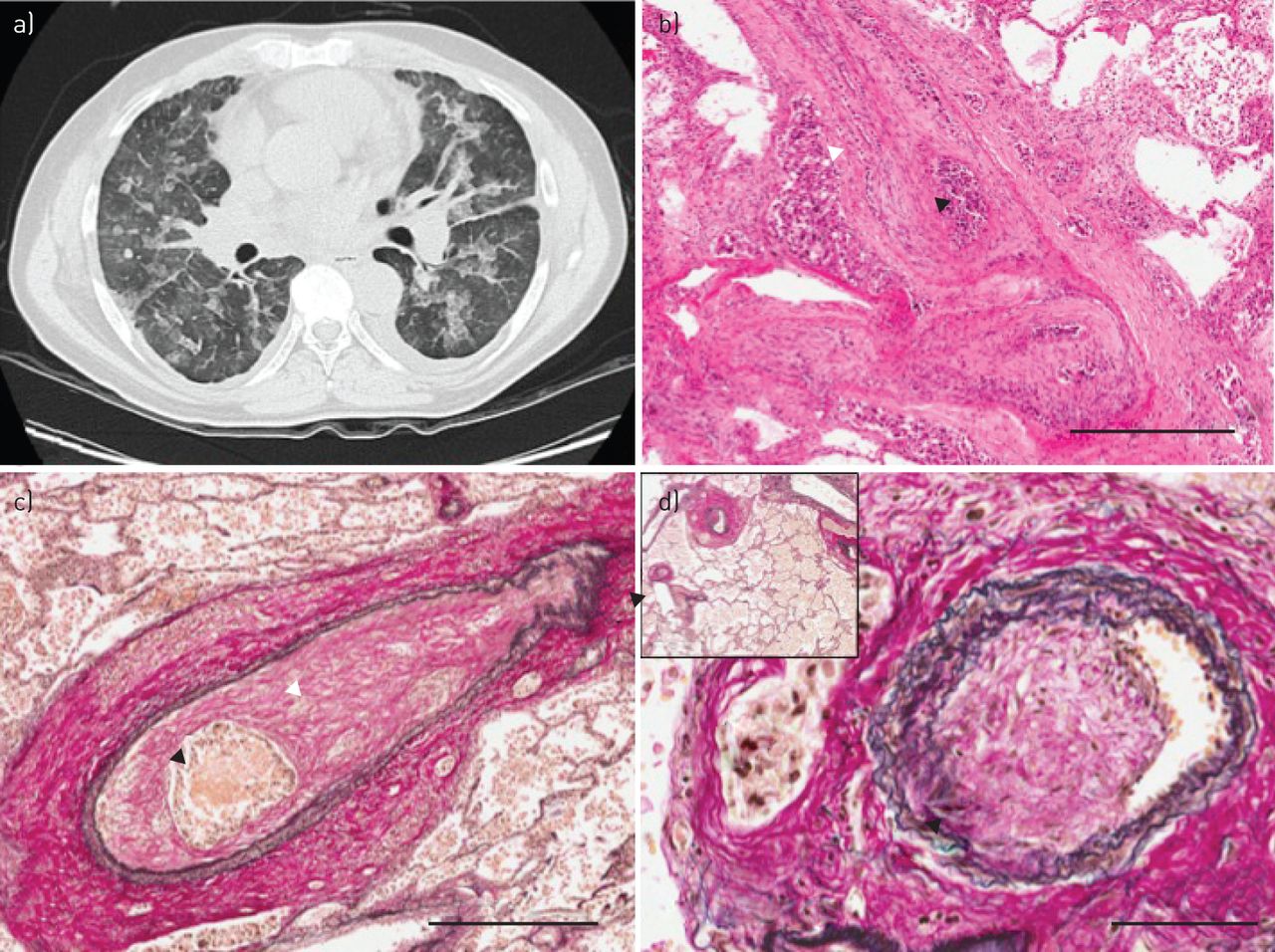
Case 1 was a 58-year-old male smoker who presented with a short history of progressive breathlessness. Initial imaging at his local hospital was suggestive of an interstitial lung process with associated PH, and he was transferred to our hospital for further assessment. Computerised tomography (CT)-pulmonary angiography (PA) excluded pulmonary embolism, and high-resolution cuts revealed widespread ground glass changes and interlobular septal thickening, consistent with interstitial and airspace oedema, rather than an interstitial lung process (figure 1a). Echocardiography revealed a moderately hypertrophic right ventricle with reduced longitudinal systolic function and an estimated right ventricular systolic pressure (RVSP) of 68 mmHg. The left ventricle was hypertrophic with good systolic function. Subsequent right and left heart catheterisation confirmed increased pulmonary vascular resistance (5.2 Wood units), with a mean pulmonary artery pressure of 40 mmHg and a mean wedge pressure of 14 mmHg. He developed progressive cardiorespiratory failure, unresponsive to a variety of treatments including diuresis and careful initiation of the phosphodiesterase inhibitor sildenafil. Post mortem analysis revealed tumour embolism in the pulmonary arteries and arterioles with severe fibrointimal arterial thickening, consistent with PTTM (figure 1b). Tumour embolism was also seen in the lymphatic vessels and veins, with reactive fibrointimal thickening and occlusion of these vessels. Immunohistochemistry was suggestive of a metastatic adenocarcinoma, consistent with a gastrointestinal primary.
{kind=link}
a) High-resolution computed tomography scanning (case 1, 14 days prior to death) showing widespread ground glass opacification, faint nodules, thickening of the interlobular septa and small bilateral pleural effusions. b) Post mortem section with occlusion of medium-sized pulmonary arterial lumen by fibrointimal proliferation (of fibroblasts and collagen) (white arrowhead) and tumour emboli (black arrowhead) (haematoxylin and eosin, 4.3× magnification, scale bar=500 µm). c) Post mortem section showing a medium-sized pulmonary artery with two elastic layers, with a normal-sized smooth muscle layer. There is exaggerated luminal occlusion by fibrointimal thickening (white arrowhead) surrounding nests of tumour emboli (black arrowhead). The adventitia contains lymphatic tumoural thrombi. Increased alveolar macrophages are seen in surrounding lung (elastic Van Gieson stain, 4.6× magnification, scale bar=500 µm). d) There was also evidence of fibrointimal proliferation within the lumen of small pulmonary veins (black arrowhead) and tumour involvement of accompanying lymphatic vessels (white arrowhead). Inset: veins close to the centrilobular bronchovascular bundles show eccentric fibrointimal remodelling (elastic Van Gieson stain, 3.4× magnification, scale bar=50 µm). Of note, in the sections we observed, pulmonary arterioles (80–100 µm external diameter) did not demonstrate significant vascular remodelling (elastic Van Gieson stain, 28.4× magnification).
Case 2 was a 42-year-old female non-smoker presenting with a short history of breathlessness and intermittent cough, unresponsive to antibiotics and steroids in the community. Initial assessment was again suggestive of a possible interstitial process associated with PH. CTPA demonstrated enlarged central pulmonary arteries and right heart chambers compatible with PH; with a mosaic attenuation pattern reflective of small vessel vasculopathy, but no evidence of pulmonary embolism. Echocardiography demonstrated severe PH (RVSP 63 mmHg) and a severely dilated right ventricle with severely reduced systolic function. She developed rapidly progressive cardiorespiratory failure, precluding invasive assessment and was unresponsive to a variety of treatments, including sildenafil and anticoagulation. Post mortem examination revealed diffuse tumour embolism throughout the pulmonary arteries (figure 1c), with marked fibrointimal thickening, consistent with PTTM. There was also evidence of fibrointimal changes in small veins and tumour involvement of accompanying lymphatic vessels (figure 1d). The tumours were a squamous carcinoma of unknown primary.
PTTM was first described in 1990 by Von Herbay et al. [2]. Histologically, PTTM is characterised by fibrocellular intimal proliferation of small pulmonary arteries and arterioles, in association with non-occlusive microscopic tumour emboli. Pulmonary tumour embolism (PTE) is a condition related to PTTM, but is quite a distinct pathophysiological process. In PTE, tumour cells metastasise to the pulmonary vasculature and trigger the local coagulation system, resulting in clot formation. The resultant increase in pulmonary vascular resistance occurs due to mechanical vessel obstruction, with no histological changes in pulmonary vessel architecture. PTE could be classifiable within the WHO PH classification system as “tumoural obstruction”, under group 5.4.
In PTTM, the development of PH is in part the consequence of remodelling of the pulmonary vasculature, rather than pure mechanical obstruction by tumour cells. However, as demonstrated in these cases, the pathological changes in PTTM can occur both within pre-capillary and post-capillary vessels (as well as pulmonary lymphatic vessels). Therefore, it could be argued that this condition can straddle several classifications: apart from fitting “by convenience” into Group 5 (unexplained multifactorial mechanisms) or perhaps Group 1′ (pulmonary veno-occlusive disease (PVOD)), given the clear clinical and histological involvement of post-capillary veins and lymphatic vessels, there is a strong analogy with the pathology observed in Group 4 (chronic thromboembolic pulmonary hypertension (CTEPH)), especially in relation to distal CTEPH. Remodelling of pulmonary veins in patients with residual CTEPH following endarterectomy surgery or in patients with distal CTEPH has indeed been recently reported [3]. Of note, we believe that this is the first time that involvement of the post-capillary vasculature has been described in the literature in the setting of PPTM.
The molecular mechanisms underlying the changes in vessel morphology associated with PTTM remain unclear. Despite this, there appears to be an intimate association between nests of tumour cells and surrounding fibrointimal proliferation in both arteries and veins (figure 1b–d). Secretion of local growth factors by trapped tumour cells may trigger vascular remodelling. Indeed, immunohistochemical studies implicate vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), tissue factor and osteopontin [4]. VEGF is a pro-angiogenic factor upregulated in the majority of human tumours, especially gastric carcinoma. Furthermore, over 50% of cases of PTTM are associated with a gastric malignancy [5]. Clearly to differentiate between PTE and PTTM, in terms of underlying mechanisms, would require more extensive clinical and biological studies.
Whilst not directly relevant to the cases described here, it is important to briefly mention other possible mechanisms involved in tumour-related PH. These include: 1) direct tumour obstruction to both large pulmonary arteries and veins; 2) chemotherapy-induced PH, both pre-capillary e.g. with dasatanib [6] and post-capillary (PVOD), e.g. with cyclophosphamide [7]; 3) chemotherapy- and radiotherapy-induced left heart dysfunction; and 4) hypoxia-related chemotherapy- and radiotherapy-induced lung damage.
Diagnosis of PTTM is notoriously difficult and requires a high index of suspicion. Uruga et al. [4] reviewed 30 cases of PTTM in 2215 consecutive autopsy cases of carcinoma of various types including gastric and lung primaries. No specific features at clinical presentation or imaging (with CT, nuclear medicine or 18 F-fluorodeoxyglucose-positron emission tomography (18F-FDG-PET) scanning) reliably diagnosed PPTM [4]. Perfusion scanning mostly showed multiple small perfusion defects, and 18F-FDG-PET showed multifocal abnormal FDG-uptake in areas of PPTM [4, 8], which may be useful although non-specific. Plasma levels of D-dimer or fibrin degradation products are, however, usually elevated [4], and could raise the suspicion of microvascular thrombosis, once larger pulmonary emboli are excluded.
Definitive diagnosis requires histological demonstration of tumour cells within the pulmonary vasculature, associated with architectural changes in pulmonary vessels. The cases diagnosed ante mortem in literature were all identified by CT-guided, surgical or transbronchial lung biopsy [9–11]. Tumour cells in the pulmonary bed may also be demonstrated by cytological analysis of blood aspirated from the wedge position during right heart catheterisation [12].
The prognosis of PTTM is extremely poor, with a median (range) survival following oxygen supplementation of 9 (1–69) days in one study [4]; ante mortem diagnosis was achieved in five out of 30 patients. Three received chemotherapy, two of whom survived several months with an associated improvement in CT findings. Interestingly, a recent case report demonstrated some improvement in PH associated with PTTM in a patient treated with imatinib, a tyrosine kinase inhibitor, which inhibits the PDGF pathway [13].
The two cases presented here highlight PTTM as a rare, likely under-diagnosed cause of PH, which is related to, but distinct from, PTE. The complex involvement of pre and post capillary vessels, as well as lymphatic vessels, may make PTTM unclassifiable in the current classification system. Despite this, it is essential that awareness is increased both to enable earlier diagnosis and to understand underlying pathogenetic mechanisms.
Relatives of both patients gave written informed consent for publication.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received January 13, 2015.
- Accepted June 2, 2015.
- Copyright ©ERS 2015










