





Abstract
Cystic fibrosis airways are frequently colonised with fungi. However, the interaction of these fungi with immune cells and the clinical relevance in cystic fibrosis lung disease are incompletely understood.
We characterised granulocytes in airway fluids and peripheral blood from cystic fibrosis patients with and without fungal colonisation, non-cystic fibrosis disease controls and healthy control subjects cross-sectionally and longitudinally and correlated these findings with lung function parameters.
Cystic fibrosis patients with chronic fungal colonisation by Aspergillus fumigatus were characterised by an accumulation of a distinct granulocyte subset, expressing the HIV coreceptor CXCR4. Percentages of airway CXCR4+ granulocytes correlated with lung disease severity in patients with cystic fibrosis.
These studies demonstrate that chronic fungal colonisation with A. fumigatus in cystic fibrosis patients is associated with CXCR4+ airway granulocytes, which may serve as a potential biomarker and therapeutic target in fungal cystic fibrosis lung disease.
Abstract
CXCR4+ airway granulocytes as a potential biomarker and therapeutic target in fungal cystic fibrosis lung disease http://ow.ly/KPdV0
Introduction
Cystic fibrosis lung disease is characterised by chronic bacterial infection [1, 2], fungal colonisation, particularly by Aspergillus fumigatus and Candida albicans [3], and airway inflammation, maintained by a continuous and nonresolving recruitment of granulocytes into the airways [4–6]. A. fumigatus is frequently found in the respiratory tracts of cystic fibrosis patients, but beyond allergic bronchopulmonary aspergillosis (ABPA), its clinical relevance in cystic fibrosis lung disease is poorly understood [3, 7–9]. Recent clinical observations support the notion that A. fumigatus airway colonisation in the absence of ABPA, a condition that has been termed “Aspergillus bronchitis” [10], may have a harmful effect on the outcome of cystic fibrosis lung disease. This relationship remains a matter of debate, but is corroborated by studies in cystic fibrosis and asthma patients that demonstrate that A. fumigatus colonisation [11] and/or sensitisation [12, 13] affect the course of lung disease, as measured by hospitalisations and lung function parameters. Other studies found more bronchiectasis, but no decline in pulmonary function in cystic fibrosis patients colonised with A. fumigatus compared to noncolonised patients [14]. Despite these observations, the conditions and underlying mechanisms by which A. fumigatus modulates cystic fibrosis lung disease remain incompletely defined.
Granulocyte recruitment and homing are tightly regulated by chemokines, which act through seven-transmembrane G protein-coupled chemokine receptors [15, 16]. Granulocyte products, such as neutrophil elastase, cause pulmonary tissue remodelling and immune receptor damage and have been shown to predict the development of bronchiectasis in cystic fibrosis patients [17]. Consequently, therapeutic interference with granulocyte recruitment represents a promising approach in cystic fibrosis lung disease [18]. We [19] and others [20, 21] have shown in previous studies that granulocytes in the airways of cystic fibrosis patients are unique in their phenotype as they are activated and characterised by a loss of the CXCL8 receptor CXCR1 (interleukin (IL)-8RA) [22], whereas noncanonical chemokine receptors, particularly CXCR4 (CD184), are upregulated on their cell surface. While CXCR1 has been shown to be involved in antibacterial host defence in cystic fibrosis lung disease [22], the clinical and functional relevance of CXCR4-expressing granulocytes in patients with cystic fibrosis lung diseases remained poorly understood.
We hypothesised that A. fumigatus colonisation in cystic fibrosis triggers innate immune responses, characterised by an accumulation of distinct airway granulocytes which modulate pulmonary disease. Our studies demonstrate that a noncanonical phenotype of granulocytes, expressing the HIV coreceptor CXCR4, accumulates in the airways of cystic fibrosis patients with A. fumigatus colonisation, and is correlated with pulmonary function in cystic fibrosis.
Material and methods
Patient cohorts
The cystic fibrosis group included 19 male and 21 female patients with a mean±sd age of 23±14 years (see table 1 for patients’ details). Inclusion/eligibility criteria were the diagnosis of cystic fibrosis by clinical symptoms and positive sweat tests (sweat Cl− concentration >60 mmol·L−1) or disease-causing mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, forced expiratory volume in 1 s (FEV1) >30% predicted and being clinically stable and on steady concomitant therapy ≥4 weeks prior to the study. 20 cystic fibrosis patients were ΔF508 homozygous, 13 were ΔF508 heterozygous carriers of the CFTR gene and 7 had CFTR mutations other than ΔF508. Bacterial and fungal species were analysed using culture-based methods and selective media in the corresponding airway fluid samples where CXCR4-expressing granulocytes were quantified using flow cytometry, and pulmonary function testing was performed. Chronic bacterial and fungal colonisation were diagnosed using the Leeds criteria [23] if the organism was present in >50% of patient samples in the year prior to analysis. 10 subjects without pulmonary diseases were selected as the control group. These subjects had no suspected or proven pulmonary disease and were free of respiratory tract infections. Eight non-cystic fibrosis lung disease controls were included. These adult patients suffered from bronchiectasis with chronic neutrophilic bronchitis, but without cystic fibrosis disease characteristics or positive sweat tests.
Characteristics of subjects in the three patient groups
For longitudinal follow-up studies, five cystic fibrosis patients with A. fumigatus colonisation and five cystic fibrosis patients without A. fumigatus colonisation, matched for age and lung function prior to the longitudinal follow-up and negative for C. albicans were studied with follow-up intervals/time points between 6 and 8 months. At each follow-up time point, percentages of CXCR4+ granulocytes in sputum, lung function (FEV1 and maximum expiratory flow at 25% of forced vital capacity (MEF25)) and IgG specific to A. fumigatus were analysed. Cystic fibrosis patients with A. fumigatus colonisation: n=5, mean±sd age 22±5 years, mean±sd FEV1 73±6% pred, median (range) FEV1 75% pred (66–82% pred); cystic fibrosis patients without A. fumigatus colonisation: n=5, mean±sd age 23±4 years, mean±sd FEV1 73±10% pred, median (range) FEV1 74% pred (63–84% pred). None of the included patients had ABPA.
This study was conducted in accordance with the amended Declaration of Helsinki. The local independent ethics committees (University of Tübingen, Tübingen, Germany and University of Munich, Munich, Germany) approved the protocol, and written informed consent was obtained from all patients.
Sample collection and processing
Induced sputum [24] and bronchoalveolar lavage fluid (BALF) were obtained, processed and stored as described previously [19, 22, 25, 26]. Cell-free sputum supernatant was stored at −80°C until analysis. The obtained BALF was filtered through two layers of sterile gauze. The first fraction of BALF was used because it contains higher percentages of granulocytes compared to the pooled fraction. Sample processing was performed immediately on ice. After centrifugation (200×g for 10 min) the supernatant was stored at −80°C until analysis. The cell pellet was resuspended in 5 mL of PBS and used for the preparation of cytospin slides and flow cytometry.
Flow cytometry
Percentages of CXCR4+ granulocytes were quantified using flow cytometry based on a modified flow cytometric strategy described previously [19], as depicted in online supplementary figure S1. Prior to staining, Fc block was used to saturate nonspecific binding sites. For staining, cells were incubated with monoclonal antibodies for 40 min, washed twice and analysed using flow cytometry [19]. For analysis, cells were first gated based on their forward scatter and side scatter characteristics. In the next step, dead cells in sputum/airway fluid samples were excluded by using propidium iodide staining. Within viable cells, granulocytes were identified by the expression of CD15. Fluorescence minus one controls were used to define gating boundaries. The manufacturers of the reagents were as follows. Antihuman CXCR4 antibody (clone: 12G5): BD Pharmingen (Heidelberg, Germany); anti-mouse Cxcr4 antibody (clone: L276F12): BioLegend (San Diego, CA, USA). All other fluorescence-activated cell sorting (FACS) reagents were from BD Pharmingen or Immunotech (Marseille, France). Calculations were performed using CellQuest analysis software (Beckton Dickinson, Erembodegem-Aalst, Belgium).
ELISA
CXCL8 and CXCL12 protein levels were measured in triplicate using a commercially available sandwich ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions in a subgroup of cystic fibrosis patients (n=30). Serum was obtained from venous blood by centrifugation at 1000×g for 10 min after blood clotting. Aliquots of serum were stored at −20°C. Specific IgE and IgG antibodies to A. fumigatus were analysed using an Immuno Cap system(Thermo Fisher/Phadia, Uppsala, Sweden) [27].
Statistical analysis
Differences between the patient groups were calculated using the nonparametric Kruskal–Wallis test. When a significant difference was found, the nonparametric Mann–Whitney U-test was applied for two-group comparisons. Correlations were verified using the Spearman ρ test. A p-value <0.05 was considered to be significant. Statistical analysis was performed using Prism 5.0 (GraphPad Software, San Diego, CA, USA) and STATA version 8.2 for Windows (STATA Corporation, College Station, TX, USA).
Results
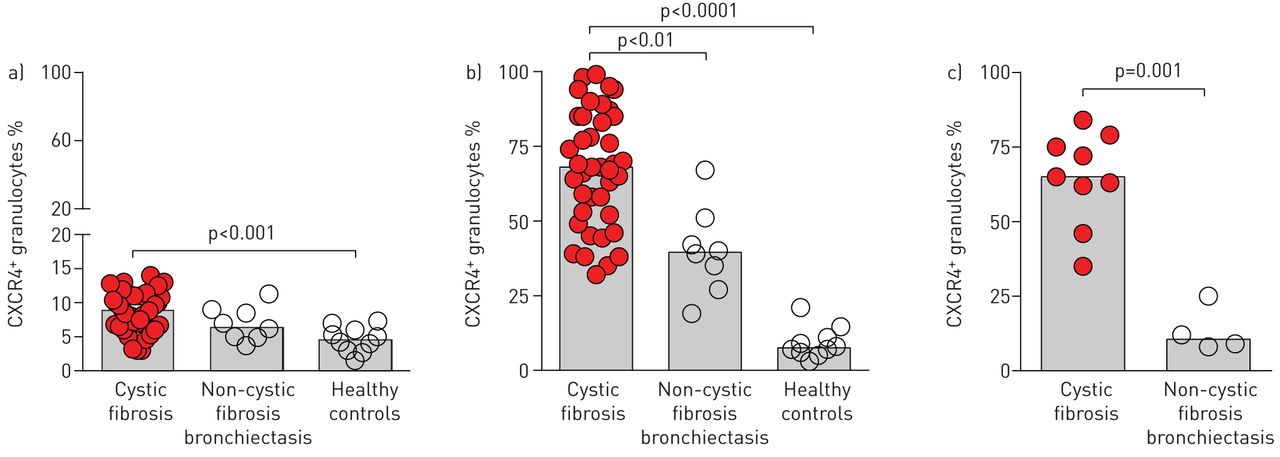
Accumulation of CXCR4+ granulocytes in cystic fibrosis lung disease
Inspired by our previous finding that CXCR4+ granulocytes accumulate in chronic lung diseases [19], we systematically quantified percentages of CXCR4+ granulocytes in peripheral blood (fig. 1a) and airway fluids (induced sputum (fig. 1b) and BALF (fig. 1c)) from cystic fibrosis patients, healthy controls and non-cystic fibrosis lung disease control subjects. These studies demonstrated that percentages of CXCR4+ granulocytes were increased in cystic fibrosis patients both in peripheral blood (fig. 1a) and to an even greater extent in airway fluids (fig. 1b and c). Percentages of CXCR4+ granulocytes showed a positive correlation with the percentage of neutrophils in cystic fibrosis airway fluids (p<0.01 for sputum and p<0.01 for BALF, assessed using cytospin staining), while no correlations were found for airway eosinophils (p>0.05), suggesting that CXCR4+ granulocytes resemble neutrophilic granulocytes in cystic fibrosis airways. While percentages of CXCR4+ granulocytes in peripheral blood from cystic fibrosis patients overlapped greatly with healthy and non-cystic fibrosis disease control subjects and only a subgroup of cystic fibrosis patients showed higher percentages of CXCR4+ granulocytes in peripheral blood compared to control subjects (fig. 1a), percentages of CXCR4+ granulocytes in both induced sputum (fig. 1b) and BALF (fig. 1c) were distinctively higher in cystic fibrosis patients compared to healthy controls, and were also increased compared to non-cystic fibrosis disease controls (fig. 1b and c). However, within the cystic fibrosis patient cohort, percentages of CXCR4+ granulocytes in both peripheral blood and airway fluids showed a wide range of values, suggesting that cystic fibrosis disease-associated factors modulate percentages of CXCR4+ granulocytes in cystic fibrosis lung disease.

CXCR4+ granulocytes in cystic fibrosis patients and controls. Percentages of CXCR4+ granulocytes were quantified by flow cytometry in a) peripheral blood, b) sputum and c) bronchoalveolar lavage fluid from cystic fibrosis, healthy control and non-cystic fibrosis bronchiectasis disease control subjects. The scattergraphs depict individual patients and vertical bars represent medians.
CXCR4+ airway granulocytes reflect fungal colonisation in cystic fibrosis lung disease
In order to dissect the cystic fibrosis-related factors that modulate percentages of CXCR4+ granulocytes in cystic fibrosis airways, we performed correlation analyses, which showed that percentages of CXCR4+ granulocytes in cystic fibrosis airway fluids correlated with colonisation by A. fumigatus (p<0.01), but not by other cystic fibrosis-related bacterial or fungal species. Stratifying the included cystic fibrosis patients in patients colonised with A. fumigatus (fig. 2a–c) or C. albicans (fig. 2d–f), these analyses further demonstrated that percentages of CXCR4+ granulocytes in the peripheral blood of cystic fibrosis patients did not differ significantly between A. fumigatus colonised or non-colonised cystic fibrosis patients (fig. 2a), whereas percentages of CXCR4+ granulocytes in induced sputum (fig. 2b) or BALF (fig. 2c) were significantly increased in cystic fibrosis patients colonised by A. fumigatus compared to non-colonised patients. In contrast to A. fumigatus, colonisation by C. albicans was not associated with percentages of CXCR4+ granulocytes in peripheral blood (fig. 2d), induced sputum (fig. 2e) or BALF (fig. 2f). Consistent with our findings in cystic fibrosis patients, airway fluid CXCR4+ granulocytes were also increased in A. fumigatus-colonised non-cystic fibrosis bronchiectasis patients compared to non-colonised patients (fig. 2b), whereas no differences were found for C. albicans (fig. 2d–f).

CXCR4+ airway granulocytes and fungal colonisation in cystic fibrosis (CF) patients. Percentages of CXCR4+ granulocytes were quantified by flow cytometry in a, d) peripheral blood, b, e) sputum and c, f) bronchoalveolar lavage fluid from CF patients and non-CF bronchiectasis disease controls with (+) or without (−) a–c) chronic Aspergillus or d–f) Candida colonisation and healthy control subjects (a, b, d, e). CF and non-CF bronchiectasis disease control patients were stratified according to fungal colonisation independently of any other pathogens detectable in CF airway fluids. The scattergraphs depict individual patients and vertical bars represent medians.
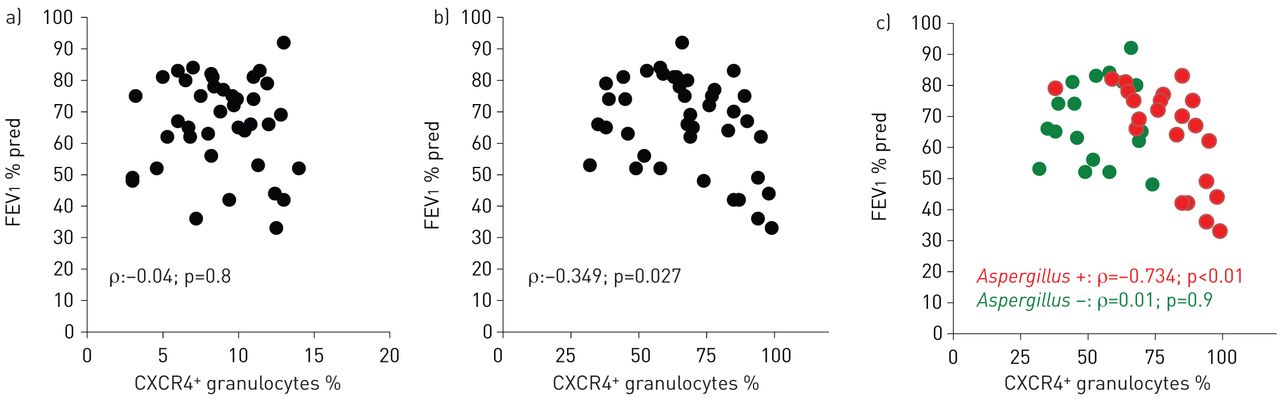
CXCR4+ airway granulocytes correlate with lung function in cystic fibrosis
Next, we analysed whether percentages of CXCR4+ granulocytes in peripheral blood or airway fluids were correlated with clinical parameters of cystic fibrosis lung disease. These studies demonstrated that percentages of CXCR4+ granulocytes in airway fluids, but not in peripheral blood, correlated inversely with pulmonary obstruction (FEV1) in cystic fibrosis patients (fig. 3a and b). Stratifying cystic fibrosis patients for A. fumigatus colonisation revealed that percentages of CXCR4+ granulocytes in airway fluids correlated inversely with lung function (FEV1) in patients colonised by A. fumigatus, but did not in non-colonised cystic fibrosis patients (fig. 3c). Percentages of CXCR4+ granulocytes in sputum correlated with specific IgG to A. fumigatus (p<0.05), whereas no correlation with specific (p>0.05) or total IgE was found (p>0.05). No correlations between antibiotics or other cystic fibrosis drugs and percentages of CXCR4+ granulocytes were found in our study cohort. Immunological analyses in a subgroup of patients further demonstrated that percentages of CXCR4+ granulocytes in airway fluids further positively correlated with protein levels of the CXCR4 ligand CXCL12 (stromal cell-derived factor (SDF)-1α) (fig. 4a) in airway fluids (induced sputum supernatants), while no correlation was found between percentages of CXCR4+ granulocytes in airway fluids and CXCL8 (IL-8) (induced sputum supernatants) (fig. 4b). Percentages of CXCR4+ granulocytes in peripheral blood did not show any correlation with protein levels of these chemokines in serum of the corresponding patients (p>0.05, data not shown).
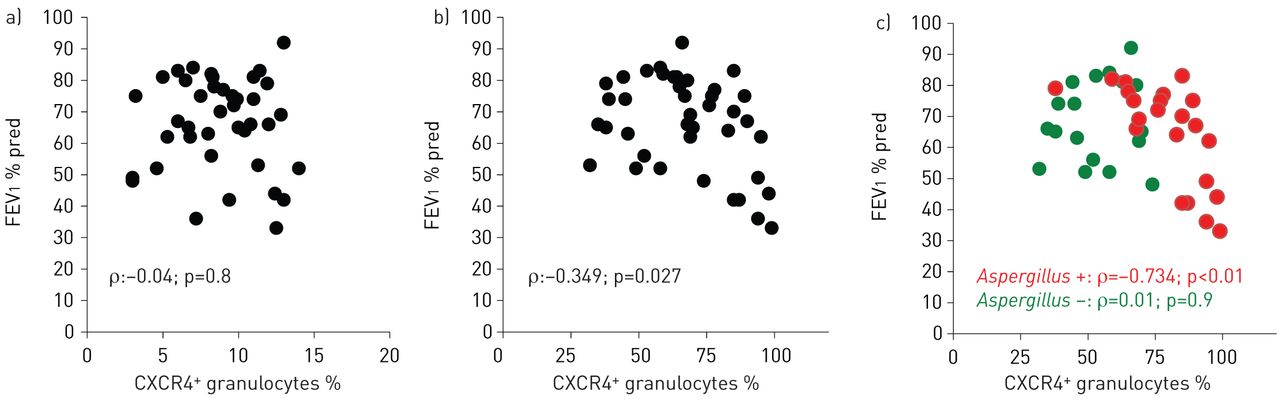
CXCR4+ granulocytes and lung function. Percentages of CXCR4+ granulocytes quantified by flow cytometry in a) peripheral blood and b, c) sputum from cystic fibrosis patients in correlation to lung function; c) cystic fibrosis patients with or without Aspergillus colonisation. FEV1: forced expiratory volume in 1 s; % pred: % predicted.

CXCR4+ airway granulocytes and chemokines. Percentages of CXCR4+ granulocytes quantified by flow cytometry in sputum from cystic fibrosis patients correlated with airway levels of a) CXCL12 and b) CXCL8. n=30.
To further understand the association of CXCR4+ airway granulocytes, A. fumigatus colonisation and lung function in the course of cystic fibrosis lung disease, we quantified CXCR4+ airway granulocytes longitudinally in five cystic fibrosis patients with A. fumigatus colonisation (fig. 5a) and five age- and lung disease severity-matched cystic fibrosis patients without (fig. 5b) A. fumigatus colonisation. No changes in diagnosis occurred in the course of these consecutive visits that may have altered patient status substantially (such as switch from Aspergillus colonisation to ABPA). Both cystic fibrosis follow-up groups were C. albicans negative. These longitudinal analyses suggested that an increase of percentages of CXCR4+ airway granulocytes over time was associated inversely with lung function (FEV1) decline in cystic fibrosis patients with A. fumigatus colonisation (fig. 5a), whereas no relationship was observed in cystic fibrosis patients without A. fumigatus colonisation (fig. 5b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Longitudinal studies. Percentages of CXCR4+ granulocytes (red lines) quantified by flow cytometry in sputum from cystic fibrosis patients, lung function (solid green lines: forced expiratory volume in 1 s (FEV1); dashed green lines: maximum expiratory flow at 25% of forced vital capacity (MEF25)) and specific IgG to Aspergillus fumigatus (blue lines) analysed longitudinally in cystic fibrosis patients a) with (n=5) or b) without (n=5) Aspergillus colonisation. Follow-up intervals between each time point were 6–8 months for all patients depicted. Data are presented as median and interquartile range. % pred: % predicted.
Discussion
Granulocytes are major determinants of airway inflammation in cystic fibrosis lung disease, but their phenotype, plasticity and clinical disease relevance remain poorly understood. We and others have described in previous studies a distinct neutrophil subtype present in cystic fibrosis airway fluids, characterised by an upregulation of the homeostatic chemokine receptor and HIV-coreceptor CXCR4 (CD184) [19, 21]. However, the pathophysiological relevance of this neutrophil phenotype has remained elusive. By quantifying CXCR4+ granulocytes in peripheral blood and airway compartments in cystic fibrosis patients as well as healthy and non-cystic fibrosis disease control subjects, we demonstrate that CXCR4+ airway granulocytes were associated with chronic colonisation by A. fumigatus in cystic fibrosis patients. Since CXCR4+ airway granulocytes also correlated with lung function, this granulocyte phenotype may represent a novel therapeutic target in fungal colonisation in cystic fibrosis lung disease.
While the role of the CXCL8 receptors CXCR1 and CXCR2 in granulocyte recruitment and functionality has been studied in greater depth in homeostasis as well as inflammatory lung diseases, such as cystic fibrosis, where CXCR1 is proteolytically disabled [22, 28–30], the role of CXCR4 on granulocytes in lung disease conditions remains rather enigmatic. Previous studies have shown that CXCR4 is stored intracellularly in granulocytes and is post-transcriptionally upregulated at the cell surface upon cell activation and/or ageing [19, 28, 31]. Further studies in mice indicated that the CXCL12 (SDF-1α)–CXCR4 axis controls the release of granulocytes from the bone marrow and their homing following senescence [32]. Another mouse study extended this view by demonstrating that the clinically-used CXCR4 inhibitor plerixafor triggers granulocyte mobilisation through a novel mechanism, involving demargination of pulmonary granulocytes and blockade of granulocyte homing to the bone marrow [33], suggesting that the pulmonary compartment represents a substantial reservoir of granulocytes, which are retained in the lung through CXCR4-mediated mechanisms. Therefore, these studies suggest that interfering with CXCR4 signalling, at least in mice, could bear the potential to promote granulocyte homing from the airways into the bone marrow. The potential function of CXCR4 in granulocyte homeostasis is further complicated by a recent study in mice demonstrating that the rhythmic modulation of the haematopoietic niche through granulocyte clearance involves CXCR4, since aged CD62LlowCXCR4high granulocytes were found to infiltrate the bone marrow and to regulate the haematopoietic niche [34]. However, the relevance of these murine findings to human conditions, particularly cystic fibrosis lung disease, remains to be established. Based on our previous experience [19] and this present study we have no evidence that CXCR4-expressing granulocytes in cystic fibrosis airways simply represent apoptotic or necrotic cells, but rather suggest that this granulocyte phenotype reflects microenvironmental adaption and cellular reprogramming, as proposed previously [20, 21, 35].
Previous studies showed that the severity of ABPA correlated with airway neutrophilia [36], which promote lung tissue damage and bronchiectasis through the release of matrix metalloproteinases [37, 38]. Studies in patients with asthma further provided evidence that A. fumigatus sensitisation was associated with airway granulocytes and was related to the course of disease [13]. The distinct association of CXCR4+ airway granulocytes with A. fumigatus, but not with other fungal or bacterial microbes, in our cystic fibrosis patient cohort, confirms and extends the importance of pulmonary granulocytes in A. fumigatus colonisation beyond ABPA. Besides these studies, murine models of fungal asthma suggest that chemokine receptors play a critical and nonredundant role in inflammatory processes and disease outcome [39, 40], but the distinct role of CXCR4 on granulocytes in the context of fungal colonisation in cystic fibrosis lung disease has not been defined so far. A. fumigatus activates the immune system through several microbial patterns and corresponding pattern recognition receptors, such as dectin-1 or toll-like receptors (TLRs) [41, 42]. Downstream responses include granulocyte recruitment and activation. Although the precise link between fungal patterns and CXCR4-expressing granulocytes remains to be established, a recent study in dendritic cells demonstrated that the fungal pathogen-associated molecular pattern and dectin-1 ligand zymosan upregulated CXCR4 surface expression on dendritic cells, thereby rendering dentritic cells susceptible to HIV infection [43]. Whether similar interactions between dectin-1 activation and CXCR4 regulation on granulocytes are operative has to be established in future studies, especially with regard to airway granulocytes in cystic fibrosis. Besides dectin-1, other pattern recognition receptors, such as specific TLRs upregulated on cystic fibrosis airway granulocytes, could further play a role in fungal granulocyte activation [22, 44].
We found in previous in vitro studies that Pseudomonas aeruginosa induced CXCR4-expressing myeloid-derived suppressor cells through a flagellin-mediated mechanism and further showed ex vivo that cystic fibrosis patients chronically colonised with this bacterium had increased myeloid-derived suppressor cells (MDSCs) (“low density” granulocytes) in peripheral blood [45]. However, in the latter study we were unable to define functional MDSCs in cystic fibrosis airways fluids due to limited sample material. Here, we quantified “high density”/conventional granulocytes in blood and airway fluids and demonstrated that A. fumigatus colonisation in cystic fibrosis patients was associated with increased percentages of CXCR4-expressing granulocytes in the airways, whereas no significant difference was found for peripheral blood. Though the relative contribution of MDSCs and granulocytes in different compartments in cystic fibrosis remains a poorly understood area of research, we speculate, based on our previous studies, that mainly P. aeruginosa and A. fumigatus modulate distinct CXCR4-expressing myeloid cell populations systemically (P. aeruginosa) or in the airways (A. fumigatus). Further studies are required to dissect the differential and compartment-specific impact of these cystic fibrosis-characteristic pathogens on MDSC and granulocyte subsets. Whether CXCR4 reflects cellular plasticity and/or granulocyte heterogeneity [46] in the cystic fibrosis microenvironment or even possesses a functional role remains to be investigated.
Since our studies showed that cystic fibrosis patients without A. fumigatus showed increased baseline percentages of CXCR4-expressing granulocytes compared to healthy control subjects, we analysed whether genetic Cftr deficiency in mice per se affects CXCR4 on granulocytes. These studies demonstrated that Cftr−/− mice showed similar percentages of CXCR4-expressing granulocytes in the bone marrow compared to age- and strain-matched Cftr+/+ mice (online supplementary fig. S2), suggesting, in line with our findings in non-cystic fibrosis bronchiectasis patients, that the genetic cystic fibrosis defect alone does not provide a sufficient explanation for increased CXCR4 expression on granulocytes. However, besides A. fumigatus, our studies failed to identify additional factors modulating CXCR4-expressing granulocytes. Consequently, A. fumigatus-independent microbial or nonmicrobial factors affecting CXCR4-expressing granulocytes in cystic fibrosis remain to be further identified.
Overall, the clinical relevance of A. fumigatus in patients with cystic fibrosis lung disease beyond ABPA remains poorly defined [3, 8, 9, 47, 48], which is, at least partially, due to the poor classification of A. fumigatus-associated disease phenotypes in cystic fibrosis. A recent study by Baxter et al. [49] addressed this issue by defining a novel immunological classification of A. fumigatus disease phenotypes in adult cystic fibrosis patients. Applying this classification to our cystic fibrosis patient cohort, CXCR4+ airway granulocytes were increased in cystic fibrosis patients colonised with A. fumigatus and with serologic signs of an antifungal immune response (specific IgG), but without serologic signs of underlying allergy or sensitisation against A. fumigatus, a phenotype most closely resembling the class 4 (Aspergillus bronchitis) subtype as defined by Baxter et al. [49] with a specific A. fumigatus IgG cut-off of 75 mg·L−1. Further studies are required to assess whether CXCR4+ airway granulocytes could serve as biomarkers and/or potential therapeutic targets in the above mentioned Aspergillus-associated disease phenotypes in patients with cystic fibrosis lung disease.
Regarding the generalisability and conclusions that may be drawn from our study, the following limitations have to be considered. 1) The limited size of the included cystic fibrosis and non-cystic fibrosis disease control populations requires further studies with larger A. fumigatus-colonised patient cohorts, particularly longitudinal studies; 2) the lack of data on CXCR4+ granulocytes in young cystic fibrosis children; and 3) the unresolved mechanistic question of how A. fumigatus modulates CXCR4 expression in cystic fibrosis granulocytes; issues that should be addressed in future studies. A further limitation is that our FACS staining panel did not include specific eosinophil markers, which precludes a precise flow cytometric discrimination of airway neutrophils and eosinophils. However, since the majority of airway cells in our cystic fibrosis airway samples were neutrophils (27-fold more neutrophils than eosinophils) and neutrophils strongly correlated with percentages of CXCR4+ airway granulocytes, while we did not find an association with airway eosinophils, we conclude that CXCR4+ airway granulocytes are unlikely to represent or contain a substantial amount of eosinophils. Nevertheless, as we cannot exclude an influence of eosinophils to our findings, we termed the CXCR4-expressing cells “granulocytes” instead of “neutrophils” in our studies.
In summary, we demonstrate that cystic fibrosis patients with A. fumigatus colonisation were characterised by an accumulation of CXCR4+ granulocytes within the airways, which correlated with the severity of pulmonary obstruction. Given that specific antagonists of CXCR4, a G-protein coupled receptor, are already in use clinically [50], targeting CXCR4+ airway granulocytes may represent a potential therapeutic approach in fungal colonisation in cystic fibrosis. Our findings in a limited number of patients with non-cystic fibrosis bronchiectasis and A. fumigatus colonisation further suggest that CXCR4+ airway granulocytes could also play a broader role in other chronic lung disease beyond cystic fibrosis, such as chronic obstructive pulmonary disease, where A. fumigatus is also found and associated with poor lung function [51].
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: Supported by the German Research Foundation (DFG, Emmy Noether Programme HA 5274/3–1 to D. Hartl), the Collaborative Research Center (SFB) 685 (D. Hartl) and the Novartis Foundation (D. Hartl). Funding information for this article has been deposited with FundRef.
Conflict of interest: None declared.
- Received September 22, 2014.
- Accepted March 19, 2015.
- Copyright ©ERS 2015
References










