





Abstract
Ageing is the main risk factor for major non-communicable chronic lung diseases, including chronic obstructive pulmonary disease, most forms of lung cancer and idiopathic pulmonary fibrosis. While the prevalence of these diseases continually increases with age, their respective incidence peaks at different times during the lifespan, suggesting specific effects of ageing on the onset and/or pathogenesis of chronic obstructive pulmonary disease, lung cancer and idiopathic pulmonary fibrosis. Recently, the nine hallmarks of ageing have been defined as cell-autonomous and non-autonomous pathways involved in ageing. Here, we review the available evidence for the involvement of each of these hallmarks in the pathogenesis of chronic obstructive pulmonary disease, lung cancer, or idiopathic pulmonary fibrosis. Importantly, we propose an additional hallmark, “dysregulation of the extracellular matrix”, which we argue acts as a crucial modifier of cell-autonomous changes and functions, and as a key feature of the above-mentioned lung diseases.
Abstract
Hallmarks of ageing are selecting factors for the pathogenesis of COPD, lung cancer and IPF http://ow.ly/HeM2n
Introduction
Societies in developed countries age at an unprecedented speed. In the country with the longest life expectancy to date, Japan, the percentage of the elderly population (aged >65 years) has increased from 5.7% in 1960 to 23.1% in 2011. Despite this dramatic increase in the elderly population, our understanding of the mechanisms of ageing on the cellular, organ and organismal level remains to be fully elucidated. Ageing is characterised as a progressive impairment of tissue/organ/organism function, resulting in increased vulnerability to environmental challenges and a growing risk of disease and death [1]. Ageing involves various heritable components, it has multiple faces and, most importantly, it serves no obvious purpose [2]. Ageing is not restricted to single cells of the organism, but affects different cell types to a variable extent and with varying impact for the overall organism [3].
One concept of ageing favours the existence of pleiotropic genes, which have opposite effects at different ages; while being beneficial at early stages, these genes are detrimental at a later age. This concept, known as the “antagonistic pleiotrophy theory”, was originally proposed by Williams [4] in 1957. To date, the p53 and mammalian target of rapamycin (mTOR) pathway have been suggested as antagonistic pleiotropic programmes [5]. Recently, also essential developmental signalling pathways, such as Wnt signalling, have been proposed to contribute to a “developmental drift” [6]. Alterations in Wnt signal activity have been linked to ageing [7–10] and aberrant activity of these pathways has been demonstrated in several chronic lung diseases (CLDs) [11, 12].
The second concept explains ageing as the “disposable soma” theory [13]. It proposes that the organism allocates its energy resources to ensure optimal growth and fertility in order to maximise its Darwinian fitness early in life. However, this growth comes at the price of insufficient maintenance and repair later in life; thereby increasing cellular damage is thus regarded as a trade-off to cell growth.
In the last years, ageing has emerged as the single greatest risk factor for chronic noncommunicable diseases, driving increased morbidity and mortality. While the above-mentioned concepts originally addressed “normal” ageing, they also provide a conceptual framework to study “abnormal” or “accelerated” ageing, which arises as a common cause for age-related chronic diseases and thus founded the concept of “geroscience” [14].
The lung is one of the few internal organs constantly exposed to the outside environment and as such the environmental impact on lung structure and function critically determines lung health [15]. The environmental challenges comprise not only active or passive cigarette smoke, but moreover indoor occupational or home exposures (e.g. fumes from cooking or heating and toxins), pollen, bacterial, or viral pathogens, amongst many others. These environmental stressors are usually cleared or counteracted by the action of specialised immune cells in the lung (such as alveolar macrophages, neutrophils or lung resident dendritic cells) and the clearing and repair capacity of structural cells, such as bronchial or alveolar epithelial cells. However, sustained or overwhelming exposure to environmental challenges, together with epigenetic changes and genetic predisposition, are most likely to tip the balance of physiological repair and renewal towards the development of CLDs (fig. 1). Notably, the impact and function of the environmental stressors, as well as organismal defence systems, is critically dependent on age in the pathogenesis of CLDs. As such, hereditary predisposing factors, the chemical (e.g. cigarette smoke) and biological nature of environmental hits (e.g. virus infection) on the various cell types of the lung including the vascular, stem and immune cell compartment may determine the outcome in terms of CLD, such as chronic obstructive pulmonary disease (COPD), lung cancer or idiopathic pulmonary fibrosis (IPF) (fig. 1).

The stochastic nature and interplay of genetic predisposition and epigenetic and environmental factors driving the development of chronic lung diseases. Genetic predisposition and epigenetic factors interact with the stochastic nature of environmental stimuli, which act on the various cell types of the lung (alveolar epithelial cells, fibroblasts, smooth muscle, endothelial cells, or immune and stem cells). The relative strength and overall integration of these challenges and predisposing factors results in the development of chronic obstructive pulmonary disease (COPD), lung cancer or idiopathic pulmonary fibrosis (IPF). Both COPD and IPF pathogenesis overlap in some instances with lung cancer development, as indicated by the arrows.
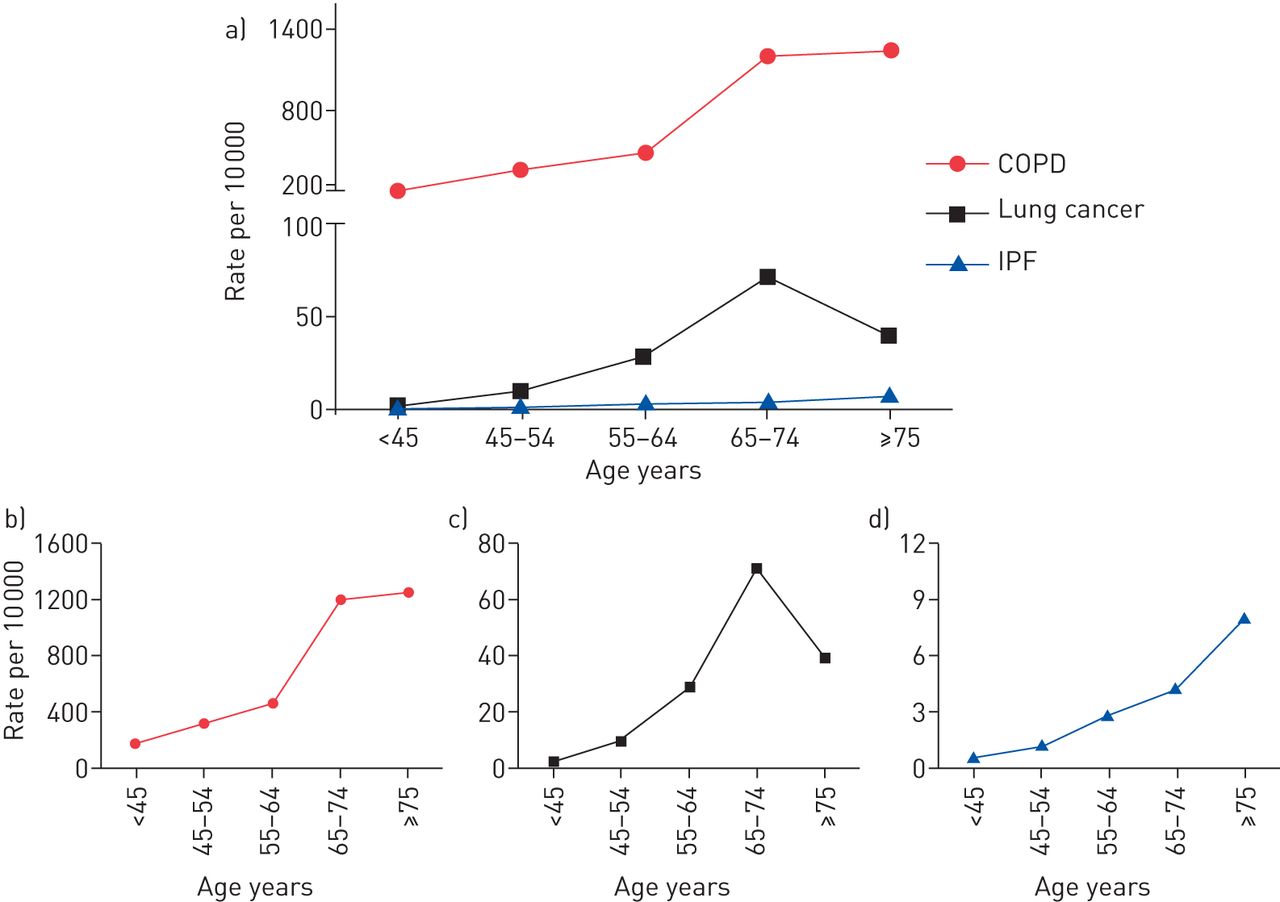
The incidence of all three CLDs dramatically rises with age (fig. 2). In 2011, chronic lower respiratory tract diseases were the third leading cause of death in people aged >65 years [19]. This health burden is also reflected by the augmented rate of newly diagnosed COPD patients, which increases from around 200 cases in 10 000 for patients aged ≤45 years to 1200 cases in 10 000 for patients aged ≥65 years (fig. 2), with the steepest increase between patients aged 65–74 years. In comparison, incidence rates for lung cancer (∼70 cases in 10 000) and IPF (∼4 cases in 10 000) are clearly lower for people aged ≥65 years. Of interest, lung cancer rates decline in the age group >75, while the number of newly diagnosed IPF cases further increases (fig. 2). These different courses of age-dependent incidence rates may reflect diverse distinct contributions of age-related mechanisms of the lung to the disease pathogenesis of COPD, lung cancer and IPF, as discussed later.
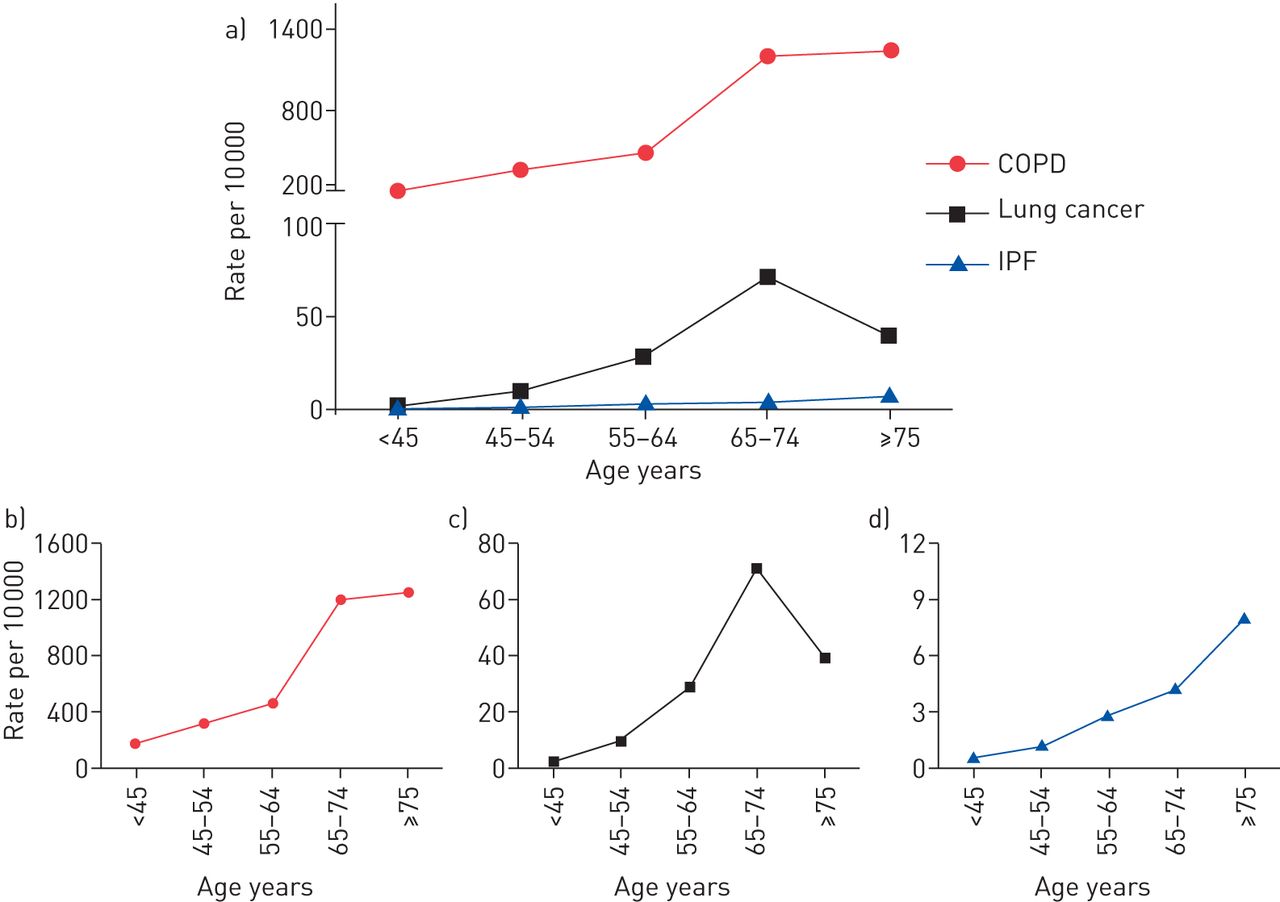
The incidence rates of chronic obstructive pulmonary disease (COPD), lung cancer and idiopathic pulmonary fibrosis (IPF) according to age in the USA. Data for age-related incidence rates (all sex) for the USA population were extracted from the following sources: COPD: [16, 17]; lung cancer: [15]; IPF: [18].
Hallmarks of ageing
In a landmark paper, López-Otín et al. [20] proposed nine hallmarks of ageing, i.e. genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, altered intercellular communication and stem cell exhaustion. Some of these hallmarks can be assigned to intrinsic properties of ageing cells. Control of cellular growth involves telomere attrition, which limits the proliferative capacity of cells due to deterioration of telomere sequences at the ends of the chromosomes [21]; the insulin growth factor 1 (IGF-1)/AKT/mTOR axis and the AMP kinase pathway that integrate cellular growth signals according to nutrient supply [22]; and mitochondrial function guaranteeing ATP production and balanced reactive oxygen species (ROS) signalling [23]. Other cell intrinsic hallmarks of ageing relate to cellular maintenance pathways: DNA damage and genomic instability is sensed by conserved DNA repair mechanisms, protein damage is controlled by the cellular proteostasis network composed of chaperones and protein degradation pathways, and cellular senescence is a cellular programme designed to shut down proliferation of unwanted and damaged cells [24–27]. Growth and maintenance pathways are tightly linked together on the molecular level by numerous regulatory cross-links and short cuts [28–30]. Another cell-intrinsic hallmark of ageing is epigenetic alterations, which can be regarded as a manifestation of persisting cellular changes to altered growth and signalling conditions [31]. Beyond these cell-autonomous alterations of different cell types within the body, there is another layer of age-related changes involving the interaction of cells with the extracellular matrix (ECM) in the tissue and intercellular communication, not only with immune but also with the stem cell compartment (fig. 3). Age-related changes in cell growth, maintenance, and epigenetics of stem cells may add to stem cell exhaustion, another hallmark of ageing, contributing to an overall impaired regenerative capacity of the organism [32, 33]. Thus, altered intercellular communication is a consequence of cell-intrinsic age-related alterations that are communicated to the surrounding cells and might result in immune cell activation. Importantly, we further propose “dysregulation of the ECM” as an additional extrinsic driving factor of ageing and will discuss the available evidence supporting this new concept (fig. 3).
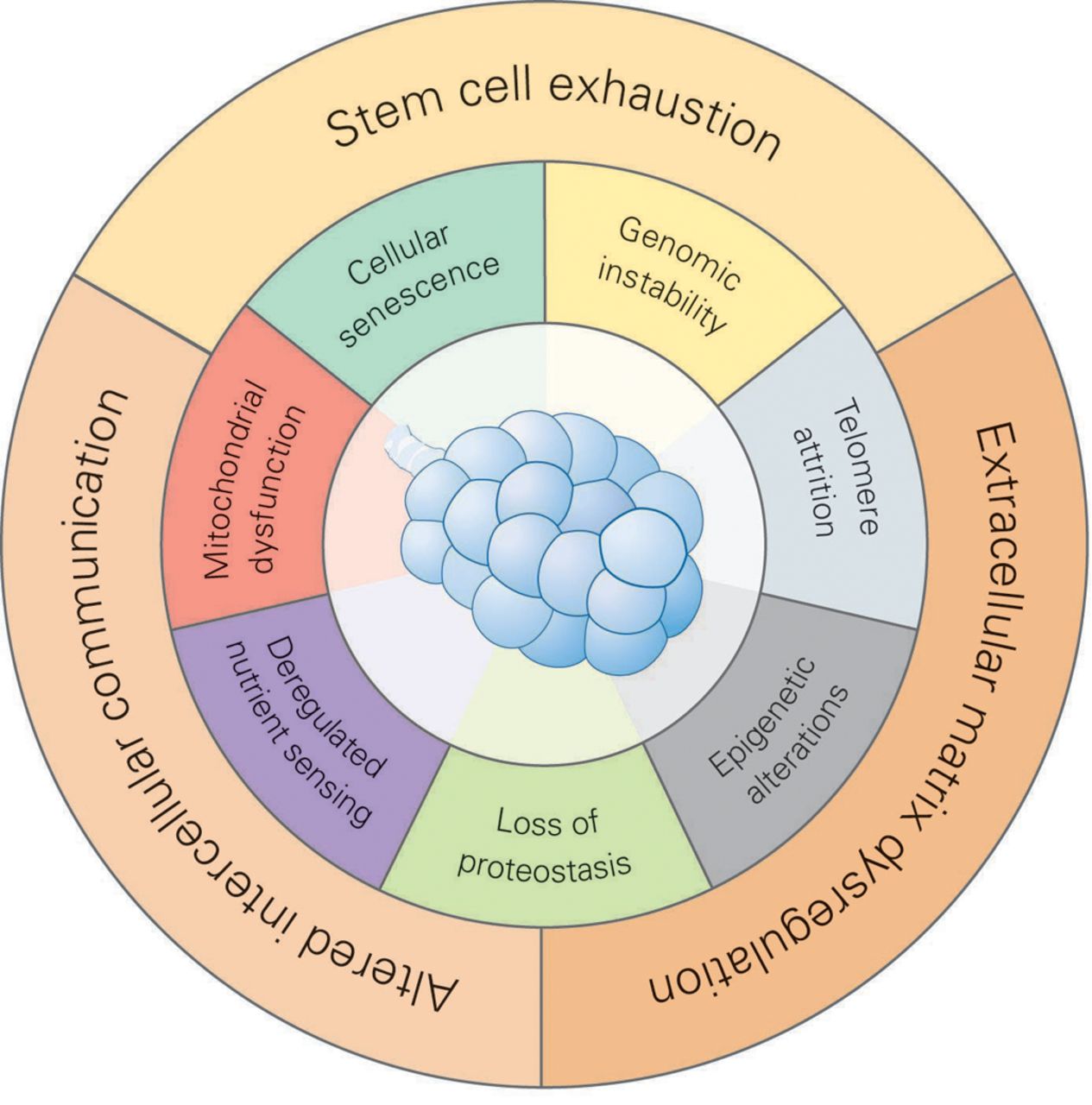
Hallmarks of the ageing lung. We propose that there are 10 hallmarks of ageing, which can be divided into cell-intrinsic hallmarks (inner circle) and cell-extrinsic factors (outer circle). The cell-intrinsic hallmarks include cellular senescence, genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, dysregulated nutrient sensing and mitochondrial dysfunction. The cell-extrinsic factors include stem cell exhaustion, extracellular matrix dysregulation and altered intercellular communication. The cell-extrinsic factors represent another layer of age-related changes that involve the interaction of cells within the tissue via the extracellular matrix or intercellular communication also with immune and stem cells. Dysregulation of the extracellular matrix is proposed as an additional hallmark of ageing for the lung.
In the following sections, we will evaluate the extended hallmarks of ageing with a particular focus on age-related CLDs, i.e. COPD, lung cancer and IPF. Affected hallmarks were selected for each disease entity on the basis of strong supporting data, which was defined as: 1) descriptive or genetic evidence from multiple clinical cohorts or at least two different laboratories; and 2) support from genetic mouse models or pharmacological targeting approaches in diseased animal models.
Hallmarks of ageing in COPD
Physiological “normal” ageing in healthy, elderly individuals contributes to a decline in lung function similar to that seen in COPD patients at Global Initiative for Chronic Obstructive Lung Disease stage I [34]. This loss of lung function is due to a variety of anatomical and physiological changes and associated with emphysema-like structural alterations of the lung. Such “senile emphysema” is characterised by alveolar enlargement in the absence of wall destruction [34] and is thus clearly different from pathophysiologically altered lungs of COPD patients. Indeed, lungs of aged rodents show similar “senile” changes when compared with lungs of young animals [35]. However, premature ageing in mouse models does not always recapitulate senile emphysema: senescence-accelerated mice (SAM), Klotho-, and senescence-marker protein-30 deficient mice show air space enlargement without airway wall destruction [36–39]. In contrast, no functional and structural changes in the lungs were observed in prematurely aged telomerase RNA knock-out mice [40]. All four premature ageing models, however, showed increased susceptibility to cigarette-smoke induced lung damage [36–40], which agrees with the notion of an age-related genetic predisposition, adding to the development of COPD. Controversial data exists whether cigarette-smoke exposure of aged mice promotes emphysema formation to a greater extent than in young animals [41].
Genomic instability
Accumulation of genetic damage over time due to genomic instability and defects in DNA repair has strongly been linked to ageing [42]. There is, however, little evidence that increased DNA damage is a driving factor for the pathogenesis of COPD and emphysema: First, deficiency in DNA repair systems as known for the Werner and Bloom progeroid syndromes, for transcription-coupled repair syndromes and according mouse models are not associated with a decline in lung function due to emphysematous changes of the lung [43]. Very similar, there are no clinical data that would suggest functional- and structural-lung alterations in syndromes of progeroid laminopathies e.g. in Hutchison-Gilford progeria syndrome [44]. Some evidence indicates that COPD patients have increased DNA-double strand breaks in lung cells and in serum. This has been linked to increased levels of oxidative stress and might contribute to increased risk for lung cancer [45–48].
Telomere attrition
The loss of protective telomere sequences at the end of chromosomes limits the proliferative capacity of cells and induces cellular senescence [21]. Loss of telomeres can be a by-product of proliferation (replicative senescence) or induced by stress [49]. For COPD, several studies shown have that both lung resident alveolar, endothelial and smooth muscle cells, as well as circulating lymphocytes, have reduced telomere lengths, which correlates directly with disease severity and smoking status [50–52]. A recent meta-analysis of 14 European studies supports the notion that reduced telomere length in circulating lymphocytes associates with COPD-related decline in lung function [53]. It also adds to the concept that accelerated ageing, as measured by telomere length, is associated with COPD and lung function decline. However, the causal contribution of telomere deficiency in the development of lung emphysema and COPD is still of debate, but has been refuelled in favour of causality by the recent identification of hereditary telomerase mutations in rare families with combined pulmonary fibrosis and emphysema syndrome [54, 55]. Telomere-deficient mice, however, do not develop spontaneous lung emphysema. However, they are more sensitive to cigarette-induced lung dysfunction [40].
Epigenetic alterations
Epigenetic alterations via DNA methylation, histone modification and noncoding RNAs have been proposed as a hallmark of ageing by which chromatin remodelling alters gene expression [20, 56]. Emerging evidence suggests that epigenetic modifications are associated with CLDs, among them COPD [57]. Circumstantial evidence indicates a role for altered DNA methylation in the pathogenesis of COPD; both, cigarette and wood smoke induce changes in DNA methylation that have been associated with an increased risk for development of COPD [58, 59]. A globally altered DNA methylation pattern has only recently been described in small airway epithelial cells [60]. Differential DNA methylation was also detected in lymphocytes of COPD patients compared to healthy controls and correlated with altered lung function and COPD severity [61]. However, whether the observed changes on DNA methylation in the lung and periphery are cause or consequence for the pathogenesis of COPD it is not yet known. To test that, animal studies using conditional knock out of DNA methyltransferases in the lung would be required. The causal contribution of histone modifications via sirtuins and histone deacetylases (HDAC) has been shown in numerous studies involving COPD patients and transgenic animals, as reviewed extensively elsewhere [57, 62, 63]. Evidence for a pathophysiological involvement of miRNAs in COPD is, however, scarce [57].
Loss of proteostasis
Accumulating damage over time also affects protein integrity and protein homeostasis, termed proteostasis. Loss of proteostasis is an early molecular event in ageing, which amplifies protein damage in the cell. It involves supervision of protein synthesis and folding as well as correct disposal of unwanted and damaged proteins via the ubiquitin-proteasome and lysosomal-autophagy pathways. Loss of proteostasis may affect specific compartments of the cell, such as the endoplasmic reticulum (ER), and induce an unfolded protein response (UPR), but it may also disturb the overall proteostasis network within the cell [64, 65]. Hereditary, as well as sporadic, alterations drive disturbed proteostasis in the pathogenesis of COPD. Hereditary mutations in α1-antitrypsin and α1-antichymotrypsin not only result in loss of protein function but misfolded proteins accumulate in the ER triggering the UPR and subsequent lung inflammation, thereby, driving chronic obstructive lung diseases [66, 67]. Loss of proteostasis is also a hallmark for sporadic cases of COPD. Reactive compounds, e.g. from cigarette smoke, oxidatively modify cellular proteins as described for histone modifying enzymes, signalling mediators and transcriptional regulators, resulting in loss of function and accelerated degradation by the proteasome [68] or autophagy pathways [69]. Cigarette smoke also induces an ER stress response by interfering with oxidative protein folding and protein function e.g. of cystic fibrosis transmembrane conductance regulator (CFTR), which may also contribute to COPD pathogenesis [70–75]. Of note, the proteasome itself is a direct target for environmental challenges as cigarette smoke and diesel exhaust have been shown to impair proteasome function [76, 77]. In accordance with that observation, proteasome function is reduced in lungs of COPD patients and in mouse models of chronic cigarette exposure, which correlates inversely with the loss of lung function [79]. However, autophagy is increased in patients with COPD and also in lungs of cigarette smoke-challenged mice [69, 79]. Only recently, selective autophagy mechanisms have also been shown to be involved in COPD-associated cilia dysfunction and mitophagy, a specialised form of mitochondrial clearance via the autophagy route [80, 81]. Dysregulated proteasome function and autophagy may shift the balance from protective proteostasis to exaggerated cellular stress responses as observed in ageing.
Deregulated nutrient sensing
Deregulated nutrient sensing in ageing involves altered growth signalling via the evolutionary conserved IGF-1/AKT/mTOR axis, which integrates signals on nutrient availability to regulate cellular growth accordingly [22, 82]. Disruption or suppression of this growth axis by caloric restriction prolongs longevity [83, 84]. Experimental data from knock-out mice indicate a possible contribution of the mTOR pathway to development of lung emphysema; while loss of the mTOR suppressor Rtp801 protected mice from cigarette-smoke induced emphysema, its lung-specific overexpression promoted lung inflammation and apoptosis of alveolar epithelial cells [85]. As mTOR signalling and autophagy, the major metabolic recycling machinery of the cell, are two sides of the same coin and intimately linked to each other, increased levels of autophagy in COPD patients may interfere with growth factor signalling via the IGF-1/mTOR axis, which remains to be investigated. Activation of the IGF-1/AKT/mTOR axis not only suppresses autophagy but also counteracts activation of forkhead box “O” (FOXO) transcription factors, which are central regulators of metabolism, stress resistance, cell cycle progression and programmed cell death [86]. Diminished expression of FOXO3 proteins has been shown in lungs of smokers and patients with COPD and of smoke-exposed mice [87, 88]. Similarly, FOXO3 ablation in mice exaggerates pulmonary emphysema in mice exposed to cigarette smoke [90]. Aberrant epidermal growth factor receptor (EGFR) signalling inhibits FOXO3A activation in COPD airways and contributes to enhanced interleukin (IL)-8 signalling [89]. Together with the recent finding that FOXO is also involved in coordinating protective autophagy, these data point towards deregulated nutrient sensing and maintenance pathways together with loss of proteostasis that contribute to the pathogenesis of chronic obstructive diseases [90].
Mitochondrial dysfunction
The contribution of mitochondrial dysfunction to ageing is not strictly dependent on mitochondrial produced ROS as originally proposed in the mitochondrial free radical theory of ageing [91]. While there is no doubt that increased ROS level and disturbed anti-oxidant protection contributes to the onset and progression of COPD [92], the exchange of mitochondrial function beyond its role as ROS generators is not yet solved. Recent data indicate that cigarette smoke alters mitochondrial structure and function but this appears to depend upon cell type and extent of damage [93, 94]. A recent study strongly indicates the disposal of smoke-damaged mitochondria via the PINK1/Parkin-mediated autophagy route as PINK1 knock-out mice were protected against mitochondrial dysfunction and smoke-induced emphysema formation [95]. Of interest, it is currently unknown whether animal models of impaired mitochondrial function develop emphysematous changes of the lung, a finding that would strongly support the contribution of mitochondrial dysfunction to COPD pathogenesis in the elderly.
Cellular senescence
Cellular senescence is a programme of defined phenotypic changes, including cell cycle arrest and senescence-mediated messaging, which prevents the propagation of damaged cells and triggers their elimination by the immune system [24, 96]. Senescence has also recently been implicated in developmental processes during tissue damage, as well as in ageing [27]. Evidence from clinical samples and mouse models demonstrates the induction of senescence in lung parenchymal cells in the progress of COPD and in response to cigarette smoke [52, 97–100]. Senescence of endothelial cells has also been linked to the pathomechanism of COPD and correlates with telomere dysfunction in these patients [50]. A recent study that used p16INK4 reporter mice to screen for age-promoting effects of environmental pollutants observed the induction of this senescence marker in response to cigarette smoke, suggesting a causal link of cigarette smoke damage and senescence induction [101]. Whether this is part of a protective response of the lung, or causally contributes to the development of obstructive lung disease requires further investigation.
Stem cell exhaustion
Endogenous stem cell exhaustion, due to repetitive and persistent injuries, has been implicated as a driving factor for several age-related diseases [102–105]. In particular, in high-turnover organs a constant asymmetric division and differentiation of stem cells is proposed to maintain tissue homeostasis and self-renewal. Over time and age, however, the demand for stem cells appears to exceed the supply provided by stem cell niches [106–108]. The lung is a slow-turnover organ [109]; however, evidence concerning human lung tissue turnover is sparse to date. Nevertheless, this notion would imply that other processes contribute to stem cell dysfunction and impaired tissue repair in the lung, in addition to an altered demand over supply ratio. In the lung, local stem cell pools have been identified in the conducting airways [110, 111], among which the best characterised are airway basal cells with the potential for self-renewal and differentiation. Recent findings further suggest that differentiated tracheal club cells (Clara cells) are able to give rise to basal cells upon injury in a genetic model of basal progenitor cell ablation [112, 113]. Their role in the development of CLDs needs to be further elucidated. In the distal lung, accumulating data demonstrates that alveolar type II epithelial cells (ATII) exhibit progenitor cell function and give rise to ATI cells [114] during development and repair, and additional potential distal stem cell populations, such as integrin α6/β4+ and surfactant protein− alveolar epithelial cells, are discussed as well [110–112].
In COPD, recent evidence suggests that basal progenitor cells, which are crucial for airway epithelial differentiation, exhibit a reduced regenerative capacity [115, 116]. Similar to other cell types, stem or progenitor cells within the lung are exposed to environmental stressors and most likely exhibit cell-autonomous hallmarks of ageing, including cellular senescence, epigenetic changes and telomere attrition [117]. Whether one or more of these mechanisms particularly affect stem cell function, compared to other cells, is currently unknown and warrants further investigation. In addition to the local stem cell pool in the lung, circulating progenitor cells have been implicated in COPD. Paschalaki et al. [118] reported that endothelial progenitor cells from smokers and COPD patients exhibit evidence of DNA damage and senescence and thus possess impaired repair function. However, others have reported decreased hematopoietic progenitors, but unchanged numbers of circulating endothelial progenitor cells in COPD patients [119, 120]. Moreover, dysfunction of other potential progenitor cell types, such as mesenchymal stromal cells (MSCs), has been associated with reduced repair capacity in a murine post-pneumectomy model [121]. Bustos et al. [122] found diminished anti-inflammatory potential of bone marrow-derived MSCs from old mice.
Altered intercellular communication
Altered intercellular communication, such as neuroendocrine dysfunction, inflammaging and immunosenescence, have been proposed as another hallmark of ageing [20]. While there is no clear link between neuroendocrine dysfunction and development or progression of chronic obstructive diseases, altered intercellular communication via the immune system is a well-established pathogenetic factor for COPD [123]. In particular, changes in the innate as well as adaptive immune responses are well characterised features of chronic obstructive diseases and emphysema and have been shown to be drivers of pulmonary emphysema in numerous animal models. While the innate immune system immediately senses environmental challenges, e.g. cigarette smoke components and smoke-induced damage of cells and the ECM, the adaptive immunity appears to propagate aberrant immune responses to acute infections or even in the absence of environmental insults [124]. Age-related changes of the immune system, commonly referred to as immunosenescence, are also held responsible for the predisposition of elderly COPD patients to acute exacerbations [125].
ECM dysregulation
ECM dysregulation is a characteristic feature of COPD. Dysregulated protease/anti-protease balance contributes to the loss of alveolar septae and emphysema formation and represents a well established paradigm for COPD pathogenesis [126, 127]. The relevance of protease imbalance for COPD pathogenesis is also reflected by hereditary deficiency of α1-antitrypsin or α1-antichymotrypsin, which not only impair cellular proteostasis but also drives emphysematous ECM remodelling in COPD [67]. Recent findings extend ECM alterations to dysregulated matrix metalloproteases (MMP) function in human COPD and cigarette smoke-induced mouse emphysema [128, 129]. In line with this, mice with reduced expression of the ECM glycoprotein fibulin-4, due to a hypomorphic fibulin-4 allele, have been shown to spontaneously develop pulmonary emphysema, as characterised by airspace enlargement and altered protease/antiprotease activity in the lung [130].
Summary
Taken together, substantial data indicate that epigenetic alterations (in particular the dysregulation of HDACs) together with loss of proteostasis, mitochondrial dysfunction, and senescence add to the pathogenesis of COPD. Additional ageing hallmarks, such as altered intercellular communication with adaptive immune responses, and abnormal ECM turnover and deposition, further contribute to COPD (fig. 4a).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hallmarks of the ageing lung in chronic obstructive pulmonary disease (COPD), lung cancer and idiopathic pulmonary fibrosis (IPF). All 10 ageing hallmarks of the lung are involved in the pathogenesis of a) COPD, b) lung cancer and c) IPF, albeit to differing extents depending on each disease. Affected hallmarks are highlighted by colour and size, while hallmarks that are not affected are shaded in white. All hallmarks were selected according to our evaluation criteria of strong supporting descriptive, mechanistic and genetic evidence from different clinical cohorts or mouse models.
Hallmarks of ageing in lung cancer
López-Ótin et al. [20] proposed cancer and ageing to be “two different manifestations of the same underlying process”, with cancer due to a gain in cellular fitness and ageing resulting from the loss of fitness. As the incidence of cancer increases with age [15], the question remains how age-related changes add to the risk of cancer development.
Genomic instability
Genomic instability is not only a hallmark of ageing but has also been considered as the major cause for neoplasia [130]. There is a clear link between impaired DNA repair and genomic instability as several hereditary disorders with mutations in genes related to DNA repair show increased susceptibility to cancer and/or to premature ageing [132]. This also applies to lung cancer. Some patients with hereditary defects in DNA repair genes, such as in Werner progeria syndrome, develop primary lung cancers [133]. Nyunoya et al. [99] showed that cigarette smoke impairs expression of Werner's syndrome protein, a RecQ helicase involved in DNA repair, which is associated with cellular senescence in lung fibroblasts and epithelial cells. Genetic variations such as single nucleotide polymorphisms (SNPs) in DNA repair genes are predictive for lung cancer progression [134, 135]. Of note, animal knock-out models of DNA repair deficiency are not particularly sensitive to lung carcinogenesis. knock-out mice of XPC, an enzyme involved in nucleotide excision repair, show only minor accumulation of mutations in the lung but frequent mutations in other tissues such as heart, liver and spleen [136]. These data suggest that the lung is less sensitive to DNA repair deficiency. Only recently, aneuploidy of nonsmall cell lung cancer (NSCLC) cells have been linked to a collapse of the nuclear envelope confirming the known link of nuclear lamina defects with failure of DNA repair as observed in nuclear laminopathies [137, 138]. Mitochondrial DNA instability has also been linked to carcinogenesis [139] but its contribution to lung tumourigenesis is not well established.
Telomere attrition
Telomere attrition plays an ambivalent role in carcinogenesis of various tumours. Shortened telomeres possibly promote early genomic instability at the onset of carcinogenesis. Age-dependent loss of telomeres may thus represent a true additive effect of ageing to carcinogenesis. However, at more advanced stages of tumour development telomere attrition opposes malignant cell growth. Thus, tumour cells repair shortened telomeres by activation of telomerases ensuring on-going proliferation of tumour cells and thus tumour progression [140–142]. In lung cancer, telomere dysfunction promotes malignant progression and metastasis in the K-ras/p53 carcinogenesis mouse model [143]. Accordingly, reduced telomere length was associated with poor prognosis of lung cancer patients [144, 145].
Epigenetic alterations
Tumour cells are generally characterised by global epigenetic alterations, such as DNA hypermethylation, altered chromatin remodelling and histone modifications, as well as deregulation of miRNAs [146]. Of note, genetic and epigenetic alterations are closely intertwined in tumourigenesis as genetic mutations disrupt the epigenome, while epigenetic processes induce genetic instability suggesting that epigenetic mutations act as “drivers” and not only “passengers” of carcinogenesis [147]. In lung cancer, epigenetic changes ranging from DNA methylation, to histone modification and miRNA dysregulation have been identified and are associated with early recurrence of lesions [148]. Several changes take place in response to cigarette smoke indicating that these changes might be driving factors for lung carcinogenesis and that epigenetic alterations might serve as early predictive markers for lung cancer [148, 149].
Loss of proteostasis
On first sight, loss of proteostasis is counterintuitive for cancer cells. As tumour cells seek to gain fitness, survival and proliferative capacity, they are dependent on proper protein function and balanced proteostasis. As malignant cells are, however, exposed to major molecular changes, due to altered metabolic demand or to stressful environments such as hypoxia or pH shifts, they need to exploit adaptive proteostasis pathways for their purpose. This has been convincingly shown for ER stress signalling and induction of the UPR, which represents an important pathway for tumour progression and survival [150, 151]. ER stress signalling has also been implicated in lung carcinogenesis [152]. Whether it acts as a friend or foe for lung cancer development is still unresolved [153]. Very similar to ER stress, autophagy functions both as a tumour suppressor pathway, to prevent tumour initiation, and as an adaptive pro-survival, to allow tumour cells to cope with metabolic and environmental stress [154]. This pivotal role has also been shown for lung cancer and challenges the application of autophagy inhibitors in cancer therapy [69, 155, 158]. The essential role of the proteasome for cell cycle control makes this a prime target for tumour therapy, as indicated by the rising numbers of proteasome inhibitors in clinical trials including lung cancer [157–159]. Elevated levels of proteasomes, therefore, represent an evasive strategy of tumour cells to chemotherapy [160–162].
Deregulated nutrient sensing
Deregulated nutrient sensing including aberrant activation of the AKT/mTOR axis ensures sustained proliferative signalling of tumour cells and thus represents a hallmark for both cancer and ageing [20, 163]. In lung cancer this pathway is generally constitutively activated by several different molecular mechanisms, as recently reviewed, making it a prime target for cancer therapy [164–166].
Mitochondrial dysfunction
Mitochondrial dysfunction has long been known as a hallmark of tumourigenesis and comprises not only altered respiratory function, described as the “Warburg effect”, but also evasion of apoptosis [163, 167]. Lung cancer is no exception from that rule [168]. Whether mitochondrial dysfunction is causal for carcinogenesis or rather the consequence of tumour cell selection and adaptation is still a matter of debate [139].
Cellular senescence
Cellular senescence represents a major protective pathway against excessive proliferation of tumour cells [169, 170]. Suppression of oncogene-induced cell senescence is thus key to sustain proliferative signalling and evade growth suppression [163]. This concept also applies to lung cancer. Several studies have established that cellular senescence is a protective mechanism for cigarette smoke induced lung cell damage which is lost upon lung cancer progression [171, 172].
Stem cell exhaustion
Stem cell exhaustion in cancer may serve as a tumour suppressor mechanism. This may account for the observed decline in lung cancer incidence rates in humans aged ≥75 years (fig. 2), which is in contrast to the continuously rising incidences for COPD and IPF. Importantly, such a decline in cancer incidence at an older age has been reported for cancer in general and was initially attributed to the selection of protected (“non-susceptible”) individuals, but may rather involve stem cell exhaustion [102, 173]. In contrast, the development of progenitor cells into cancer stem cells, which exhibit dysregulated self-renewal and differentiation is well established [163, 174, 175]. Cancer stem cells have been identified in NSCLC and represent promising therapeutic targets [178, 179]. Thus, transformation of progenitor cells by oncogenic signalling rather than stem cell exhaustion fosters carcinogenesis. Along this line, Desai et al. [178] reported that ATII stem-cell function is controlled by oncogenic EGFR-KRAS signalling, which might lead to cancer development. The precise contribution of stem cell exhaustion of cancer stem cells needs to be delineated in future studies.
Intercellular communication
It is well established that tumour progression involves altered intercellular communication that establishes a distinct microenvironment of cancer cells with various stromal cell types to support growth, angiogenesis and invasion [165, 181]. In addition, altered communication of cancer cells to immune cells allows malignant cells to escape immune surveillance, a process termed immune-evasion, which has been proposed as another emerging hallmark for tumourigenesis [163, 180]. In addition, tumour-associated macrophages have been shown to promote tumour progression by increasing cancer cell migration and invasiveness [183]. Accordingly, blocking immune checkpoints and developing vaccination strategies against cancer cells in form of immunotherapies offer exciting new treatment options also for lung cancer [184, 185]. Recent evidence suggests that immuno-ageing may also contribute to the age-related risk to develop cancer but data for lung cancer are lacking [184, 185].
ECM dysregulation
Initiation and progression of tumour growth and metastasis also involves ECM dysregulation [163]. The ECM represents a major part of the local tumour niche and has been shown to directly promote cellular transformation and metastasis, as well as neoangiogenesis and inflammation [186, 187]. The main contributors of altered activities of ECM remodelling enzymes and thus abnormal ECM metabolism are thought to be stromal cells, including cancer-associated fibroblasts, and immune cells [163, 188, 189]. Consequently, identification of tumour and metastases-associated ECM signatures using high-throughput proteomics may offer a novel concept for personalised tumour diagnosis and therapy, as also suggested for lung cancer [190, 191]. For lung adenocarcinomas, recent high-throughput analysis revealed the contribution of particular ECM molecules, such as fibronectin, as well as integrin interactions to metastatic lung cancer growth [192].
Summary
In summary, strong evidence supports that nearly all of the reported hallmarks are involved in lung cancer pathogenesis. In particular, ECM alterations have been strongly linked with the development of cancer and metastasis (fig. 4b). However, current data regarding stem cell exhaustion are conflicting, as several mechanisms, including stem cell exhaustion as an effective machinery to suppress tumour growth, as well as the development of cancer stem cells driving the disease, have been described.
Hallmarks of ageing in IPF
As the incidence of IPF continuously increases with age, ageing-related mechanisms have been proposed as a pathogenetic driver for the onset of this devastating lung disease [193–195].
Genomic instability
Some evidence indicates that genomic instability, as caused by defects in DNA repair mechanisms, may contribute to fibrotic lung diseases, since some patients with the premature ageing disease dyskeratosis congenital develop pulmonary fibrosis [196]. Secondly, the ataxia telangiectasia gene is an important sensor of DNA damage and a subset of patients with ataxia telangiectasia develop forms of interstitial lung disease [197]. Another study has indicated a putative link between genomic instability and fibrosis in two families with DNA repair deficiencies of unknown aetiology [198]. However, pulmonary fibrosis is not a characteristic feature of patients with known hereditary DNA repair or progeroid syndromes [44]. While loss of heterozygosity and microsatellite instability were detected in IPF patients, this was not related to disease severity and did not fulfil the criteria of a replication error repair phenotype [199, 200].
Telomere attrition
Telomere attrition is a driving factor of lung fibrosis. Mutations of telomerase genes, which contribute to reduced telomere length and cell growth [201], have been found in 8–15% of familial and in 1–3% of sporadic cases of pulmonary fibrosis [54, 202]. This prevalence makes pulmonary fibrosis the most common manifestation of mutant telomere genes [203–207]. Patients with mutations in telomerase and telomere genes have about 50% reduced telomerase activity, resulting in accelerated telomere loss [203]. Moreover, recent genome-wide association studies revealed that SNPs within the TERT gene increase the risk of lung fibrosis [207, 208]. A recent publication described a mutation in the TERT complex-associated dyskerin1 gene in a family of interstitial pneumonia [209]. However, mice deficient in telomere genes do not show any signs of lung fibrosis. When challenged with cigarette smoke, these mice develop emphysema, but when instilled with bleomycin, pulmonary fibrosis is either attenuated or not at all affected by telomerase deficiency [40, 210]. In addition, a telomerase activator was recently shown to suppress pulmonary fibrosis in mice [211]. As these animal data are in obvious contrast to the genetic evidence from IPF patients, they either reflect a species-specific effect of telomere attrition or point towards a more complex regulation of telomere length in pulmonary fibrosis that may also depend on the cell type affected, as recently suggested from a study on telomere activity in IPF lung fibroblasts [212].
Epigenetic alterations
Epigenetic alterations are clearly associated with the pathogenesis of IPF. In particular, altered microRNA profiles, mainly the downregulation of miRNA expression, have been observed in IPF patients [213–215]. Experimental inhibition of those miRNAs in mice induced hallmarks of fibrotic lung disease, while their overexpression protected mice from bleomycin-induced lung fibrosis [214, 215]. Epigenetic regulation via miRNA has recently been extended to the altered expression of the DNA methylase DNMT-1 in IPF [216]. In accordance with these data, there is also evidence for altered DNA methylation in IPF lungs: epigenome analysis revealed globally altered methylation patterns of hundreds of CpG islands, but also of specific promoters of genes implicated in lung fibrosis, which correlated with altered expression of the respective genes [217–221]. In addition to DNA methylation, epigenetic histone modifications, in particular those mediated by HDACs, may contribute to lung fibrosis, as they were shown to regulate expression of single target genes of pulmonary fibrosis [220, 222].
Loss of proteostasis
Loss of proteostasis with aberrant protein accumulation contributes to IPF pathogenesis, as indicated by (rare) familial cases of IPF, which carry heterozygous mutations in the surfactant protein C or A2 genes [223–227]. Several of these mutant secretory proteins fail to exit the ER but accumulate within the cell instead. As a consequence, the folding capacity of ER-resident chaperones is saturated and an adaptive UPR is launched [227]. Activation of the UPR aims to restore protein homeostasis of the ER. When this fails, e.g. upon severe or chronic ER stress, apoptosis of these cells is initiated [228, 229]. There is growing evidence for the activation of the UPR in ATII cells in sporadic cases of IPF [230–232]. Moreover, virus infection of epithelial cells, e.g. with murine gamma herpesvirus (mHPV), induces ER stress and might serve as an additional challenge to the ER, which then tips the balance towards a detrimental UPR response [233]. Infection of aged (but not young) mice with mHPV69 induced typical features of pulmonary fibrosis, supporting the notion that altered ER proteostasis in ageing contributes to pulmonary fibrosis [234]. Concerning the degradative arms of the proteostasis network, impaired activation of autophagy has been suggested to contribute to IPF pathogenesis [235, 236]. The role of proteasomal protein degradation in IPF development is unknown, but cannot be excluded as proteasome inhibitors have been shown to counteract bleomycin-induced fibrosis in mice [237].
Dysregulated nutrient sensing
The fact that IPF is a fibroproliferative disease of the lung with aberrant expression of growth factors and activation of the IGF-1/AKT/mTOR axis indicates that dysregulated nutrient sensing is involved in IPF pathogenesis [238]. Enhanced IGF-1 signalling drives pulmonary fibrosis in mice [239, 240], while blocking the IGF-1 receptor counteracted bleomycin-induced lung fibrosis in mice [241]. Accordingly, clinical trials using tyrosine kinase inhibitors to interfere with growth factor signalling [242] have been performed. Blocking the mTOR pathway by rapamycin has also been shown to attenuate transforming growth factor-β (TGF-β) induced lung fibrosis in mice [243]. In accordance with increased activation of the mTOR axis, inactivation of FOXO3A has been observed in IPF fibroblastic foci [244, 245]. Inhibition of the PI3 K/AKT signalling axis counteracted TGF-α and -β-induced lung fibrosis in mice [246, 247]. However, dysregulated nutrient sensing via the AKT/mTOR axis may only be one side of the coin and aberrant mesenchymal-epithelial crosstalk the other. This is indicated by several studies showing that enhanced growth factor signalling, e.g. by deletion of the main AKT-inhibiting phosphatase PTEN, not only drives fibroproliferation, but also regulates myofibroblast differentiation and integrity of alveolar epithelial cells [248–251].
Mitochondrial dysfunction
Mitochondrial dysfunction has been suggested as a pathogenetic factor in IPF. However, while mitochondrial ROS production is involved in profibrotic TGF-β signalling and NOX4 activation in the fibrotic lung [252–254], it remains to be investigated whether dysregulation of mitochondrial metabolism also contributes to development of pulmonary fibrosis, as suggested recently [255].
Cellular senescence
Cellular senescence is the cell's intrinsic programme to respond to replicative or stress-induced telomere shortening. Given the pathogenetic role of telomere dysfunction in IPF, it is thus not surprising that senescent cells have been detected in lungs of IPF patients. Minagawa et al. [256] observed increased staining for senescence-associated beta-galactosidase and p21 specifically in alveolar epithelial cells. In the bleomycin model of lung injury, Aoshiba et al. [257] reported a senescence-associated secretory phenotype. Similarly, pneumocyte senescence has been observed in radiation-induced lung fibrosis in mice [258]. In accordance with senescence as a driving factor for pulmonary fibrosis, reduced senescence of alveolar epithelial cells protects caveolin-1 deficient mice from bleomycin-induced lung damage and fibrosis. Similarly, bleomycin challenge of SAM prone mice enhanced development of lung fibrosis compared to the respective resistant strain [259]. This effect was mainly ascribed to an altered function of bone-marrow derived stem cells in senescence-prone mice; however, senescence of ATII cells was not investigated.
Stem cell exhaustion
IPF pathogenesis involves impaired wound repair capacity and distorted epithelial-mesenchymal crosstalk, alongside pronounced changes in the bronchial epithelium with abnormal proliferation and bronchiolisation of alveolar regions. The renewal capacity of stem cell niches in the bronchial epithelial layer is well-established [110, 111], as such, stem cell exhaustion has been proposed as a contributing factor to IPF. Alveolar epithelial cell injury and hyperplasia, in particular phenotypic changes of ATII cells, represent another key finding in IPF [260, 261]. Several lines of evidence suggest that ATII cells serve as progenitor cells for ATI cells [115] and may thus represent the prime target for age-related progenitor cell exhaustion in pulmonary fibrosis, as they exhibit several age-related cell-autonomous changes, such as dysregulated proliferative capacity, ER stress and telomere attrition [203, 234, 262]. Next to these lung-resident cells, bone-marrow derived endothelial progenitor cells are reduced in IPF patients [263]. As similar findings have been reported in COPD, the exhaustion of bone-marrow stem cell niches may be a general modifier for the development of chronic diseases during ageing, which needs further investigation.
Altered intercellular communication
To date, the causal role of altered intercellular communication in the pathogenesis of IPF is controversial with respect to the involvement of immune cells. We currently lack clear evidence linking immunosenescene to the onset or progression of pulmonary fibrosis [264, 265]. However, there is no doubt that intercellular communication is absolutely essential and a driving force for the misguided wound healing process of the alveolar epithelium that promotes pulmonary fibrosis [264, 266]. The initial damage of the alveolar epithelium by genetic predisposition (e.g. surfactant protein C mutations, which drive ER-stress-mediated apoptosis) or environmental insults triggers pulmonary repair by activation and differentiation of mesenchymal cells, which may result in interstitial lung fibrosis upon deregulation. This process involves both pro-inflammatory and profibrotic mediators for the initiation and maintenance of the fibrotic phenotype [265]. In addition, reactivation of developmental pathways, such as Wnt, TGF-β, Notch or sonic hedgehog signalling have been demonstrated to contribute to impaired cellular crosstalk and potentiate profibrotic signalling. Importantly, inhibition of these pathways attenuates bleomycin-induced fibrosis in mice [267–271]. It also involves the interaction of different cells types of the lung, but also of the immune and stem cell compartment [264].
ECM dysregulation
ECM dysregulation including distorted expression, turnover, or deposition of ECM components represents a key feature of IPF. Several factors are involved in (dys)-balancing the ECM, such as de novo synthesis and ECM deposition induced by profibrotic growth factors and proteolytic degradation by MMPs and tissue inhibitors of metalloproteinases [260, 261, 272]. Lung-specific loss of ECM components, such as α3-laminin, worsens bleomycin-induced pulmonary fibrosis, as recently shown [273]. The role of cell-matrix interactions represents an area of active investigation on the ability of lung matrix to prime cell behaviour, such as myofibroblast activation [274], but it would also impact on exogenous progenitor cell recruitment [275–277]. Along these lines, increased numbers of fibrocytes in the bone marrow, as well as in the lung, have been reported in older mice upon lung injury. Moreover, ECM secreted by lung fibroblasts from older animals further drove myofibroblast differentiation of fibrocytes in vitro [278]. Importantly, recent findings suggest that next to mesenchymal cells other structural cells, such as alveolar epithelial or circulating (progenitor) cells, contribute to ECM production [279–280]. All these cell types potentially exhibit age-related cell-autonomous alterations, as described earlier. Moreover, these cells are also most likely to change their ECM production profile with age, as reported for fibroblasts [282] and in renal fibrosis [283].
Summary
To conclude this part, substantial evidence supports the view that two key cell populations, epithelial cells and fibroblasts, exhibit strong features of ageing in IPF. These include cell-autonomous alterations, such as genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, or cellular senescence. Moreover, altered intercellular communication and stem cell exhaustion are potential hallmarks of lung fibrosis. Strong evidence further suggests that ECM dysregulation is a prominent ageing feature with significant pathological consequence, driven by impaired cell-matrix interaction (fig. 4c).
Conclusion
ECM dysregulation as a new hallmark of lung ageing
In the preceding paragraphs, we have proposed that dysregulated ECM dynamics and subsequent impaired cell/matrix interaction is an essential component in the transmission of cell-autonomous changes to the local environment for an aged organism. Dysregulated ECM dynamics, thereby, contributes directly to the development of age-related CLD, such as COPD, lung cancer, or IPF.
Alterations in the ECM are found in several organs and tissues during physiological ageing. Beyond the gain of skin wrinkles due to loss of matrix elasticity, enhanced matrix deposition and altered matrix composition have been reported for the ageing heart, kidney, skeletal muscle, and, most importantly, for the lung in humans and animal models [284–289]. With respect to lung cancer, Hanahan and Weinberg [163] emphasised the influence of the tumour microenvironment, i.e. the stroma, as one hallmark of tumourigenesis, which directly affects cancer-enabling pathomechanisms and metastasis. Moreover, the ECM within decellularised lungs of aged mice compromised proper repopulation of lung epithelial cells, similarly to emphysematous scaffolds [289]. This supports the notion that the ECM not only serves as a scaffold and essential mirror of cell-specific changes and phenotypes, but actively signals to the cell via integrins or mechanoreceptor-mediated outside-in signalling [290–292]. Together with the finding that the capacity of cells to produce and secrete distinct ECM components changes during ageing [282], these data collectively emphasise the complex interplay of any cell with its surrounding ECM. Importantly, the interaction of the ECM with secreted growth factors further affects its function as an “instructing niche” [293]. In line with this concept, Parker et al. [276] recently reported that fibrotic ECM in IPF patients clearly instructs fibroblasts to enhance ECM production. This resulted in a profibrotic vicious cycle, which further contributed to the progression and worsening of lung fibrosis.
Outlook and possible implications
Taken together, it is evident that all ageing hallmarks are involved in the pathogenesis of the age-related CLDs, i.e. COPD, lung cancer and IPF, albeit to a differing extent depending on the disease (fig. 4). While the extent and intensity of ageing-related research and thus knowledge differs between the different CLDs (currently, lung cancer is likely most extensively studied), we propose that distinct ageing hallmarks contribute to the pathogenesis of each respective CLDs, with the particular notion that dysregulation of the ECM represents a relevant novel hallmark for all of them (fig. 4).
The accumulating evidence we summarised in this review strongly suggests that ageing hallmarks are selecting factors for the pathogenesis of COPD, lung cancer or IPF. While this might imply that ageing is a driving factor for these diseases, we propose that the molecular mechanisms underlying ageing are of such central importance for organ growth and maintenance that any pathogenesis will involve deregulation of one or the other pathway. It will thus be of utmost importance for future studies in the field to extend research on the identification of therapeutically amenable maintenance mechanisms, which are central for cellular repair, control of molecular damage, as well as for stem-cell function and cell/matrix interactions in the lung. Correcting dysregulation of these central maintenance hubs will interfere with amplification and exaggeration of cellular damage as disease progresses. This may then contribute to the activation of endogenous lung repair mechanisms in order to cope with sustained environmental challenges of the lung and interfere with age-related disease progression.
Acknowledgement
We thank Frank Dahlström (Comprehensive Pneumology Center, Helmholtz Zentrum München, Munich, Germany) for his support with the figures.
Footnotes
Support statement: The authors were supported by the Helmholtz Association (Berlin, Germany). M. Königshoff is supported by an ERC starting grant 261302-PEARL. Funding information for this article has been deposited with FundRef.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received October 8, 2014.
- Accepted December 11, 2014.
- Copyright ©ERS 2015
References










