





To the Editor:
Hermansky–Pudlak syndrome (HPS) is a rare, heterogeneously inherited, autosomal recessive group of disorders presenting with a triad of oculocutaneous albinism, a haemorrhagic defect secondary to platelet dysfunction and accumulation of ceroid-like material in the reticuloendothelial system [1, 2]. Although pulmonary fibrosis has occurred in a number of patients with this syndrome and generally proves fatal in some subtypes [1, 2], there have been no reports of pneumothorax associated with HPS.
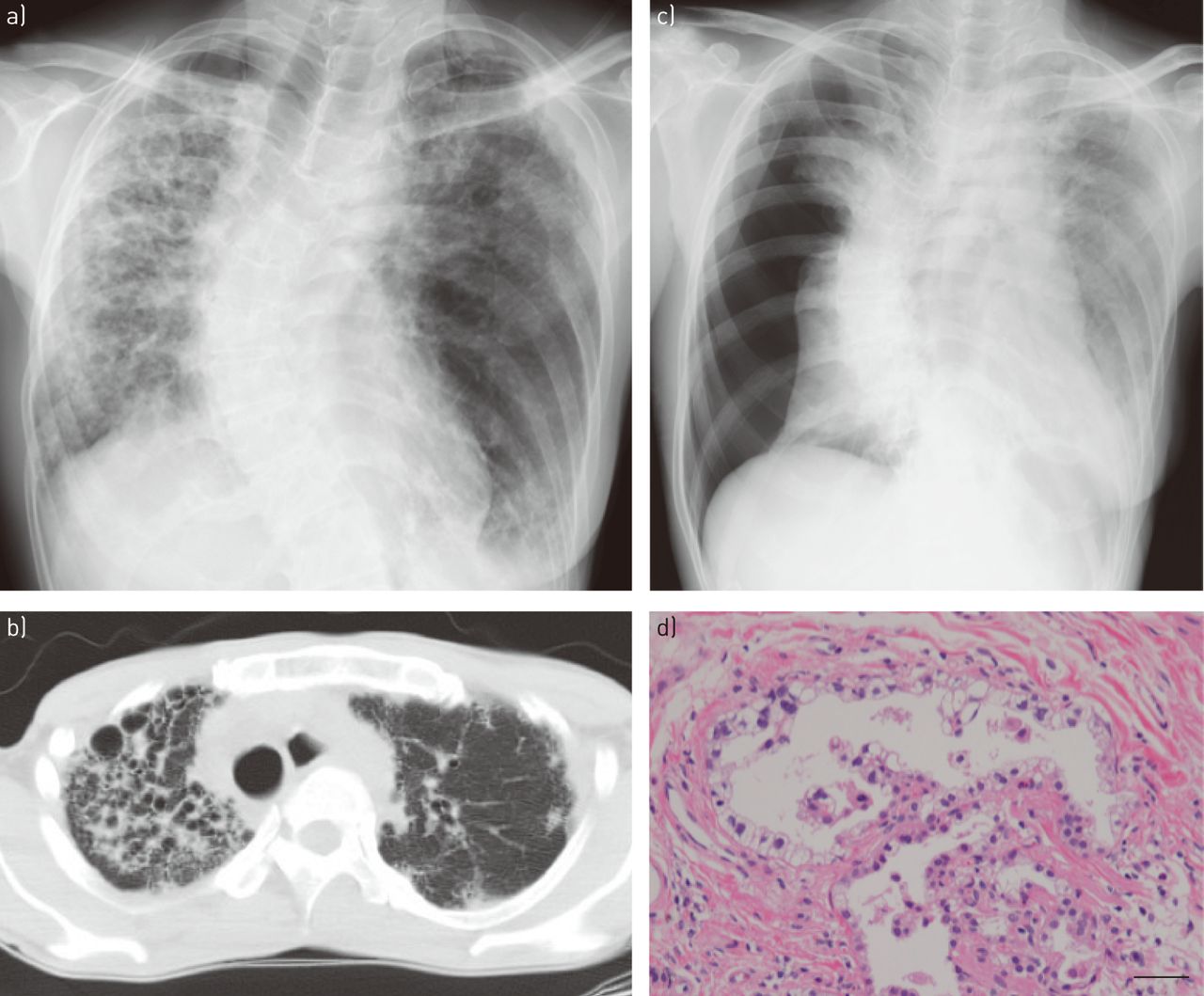
In 2006, a 49-year-old woman was referred to our hospital for detailed investigation of diffuse opacities detected by chest radiography. At the time, the patient was found to have oculocutaneous albinism, absence of platelet-dense bodies and mutations in the HPS1 gene, leading to the diagnosis of pulmonary fibrosis associated with HPS. The patient had never smoked and was a child of parents with a history of consanguineous marriage (cousins). Around 2010, chest radiography revealed bilateral reticulonodular opacities and decreased lung volumes, with right lung predominance (fig. 1a). Computed tomography showed small lung volumes, reticular opacities, airspace consolidation, diffuse traction bronchiectasis and cystic changes, mainly in the right lung (fig. 1b). The cystic changes showed a central, patchy, peribronchovascular distribution rather than being confined to the subpleural space. Pulmonary function tests showed a restrictive pattern. Oral administration of pirfenidone and home oxygen therapy were initiated, and the patient was followed up at the outpatient clinic.

{kind=link}
a, b) Radiological findings of fibrosis due to Hermansky–Pudlak syndrome (HPS). a) Chest radiography shows reduced lung volumes and reticulonodular shadows, mainly in the right lung. b) Computed tomography reveals a reticular or cystic interstitial pattern, and infiltrates, mainly in the right lung. c, d) Radiological and pathological findings of HPS at the time of occurrence of pneumothorax. c) Chest radiography shows complete collapse of the right lung. d) Prominent vacuolated pneumocytes line an alveolus. Haematoxylin and eosin staining. Scale bar=100 μm.
In 2011, the patient presented with dyspnoea of sudden onset. Chest radiography showed complete collapse of the right lung (fig. 1c), and the patient was diagnosed as having secondary pneumothorax caused by HPS. Although conservative therapy was undertaken with insertion of a chest tube, the air leak persisted, and surgery was scheduled. During video-assisted thoracic surgery, multiple bullae were observed, mainly in the apical portion of the lung. Wedge resection of the lung and ligation of the bullae were performed. The lung parenchyma showed diffuse stiffening. Abrasion of the parietal pleura was also performed. Because oxygenation could not be maintained by single-lung ventilation during the surgery, intermittent bilateral lung ventilation was adopted. Moreover, because more oozing than usual was found after the pleural abrasion, electrocautery was carefully performed. The postoperative course was favourable and no platelet transfusions were required. On histopathological examination, prominent fibrosis and a diffuse, interstitial, lymphocytic infiltrate were present. Emphysematous bullae and foamy swelling of the type II pneumocytes were also observed (fig. 1d). After 2 years of receiving home oxygen therapy, the patient died of respiratory failure at the age of 55 years while waiting for a lung transplant.
HPS is characterised by oculocutaneous albinism associated with a bleeding diathesis and pulmonary fibrosis [2]. Worldwide, it is an extraordinarily rare disorder, occurring at a prevalence of one in 500 000 to one in 1 000 000 [1]. It has been recognised as a genetically heterogeneous set of related autosomal recessive conditions due to mutations in genes that mostly function in membrane and protein trafficking [1]. People with HPS1 and HPS4 mutations develop pulmonary fibrosis, with an estimated 80% of cases with HPS1 mutations being afflicted with this condition [1, 2]. Pulmonary fibrosis is the most serious complication and main reason of death in HPS patients [2]. Based on the most prominent hypothesis of the mechanism of pulmonary fibrosis development, intracellular disruption of type II pneumocytes by ceroid may trigger an inflammation cascade, cytokine production and fibroblast proliferation, leading to progressive fibrosis [2]. Although no cases of fibrosis at sites other than the lungs in HPS patients have been reported, organs such as the lungs, intestines and kidneys of HPS patients are reported to demonstrate pulmonary fibrosis, granulomatous colitis and renal failure of varying severities, respectively, secondary to ceroid deposition [2].
Steroid therapy is not effective [2]. Lung transplantation is currently the only potentially life-extending therapy for patients with severe, progressive pulmonary fibrosis [3], although the efficacy of the antifibrotic agent pirfenidone has been investigated for the pulmonary fibrosis in HPS patients, with promising results [2].
Reports on the pulmonary pathological features of HPS are scarce [1, 4]. The fibrosis is characteristically widespread and uneven in distribution, without the localisation seen in usual interstitial pneumonia. Most characteristic is the florid foamy swelling of the type II pneumocytes, suggesting a form of cellular degeneration associated with accumulation of surfactant [4]. The active fibroblastic foci seen in usual interstitial pneumonia are generally not present in HPS [4].
To the best of our knowledge, ours is the first case report, in the English literature, of surgical treatment of pneumothorax associated with HPS. Although secondary pneumothorax occasionally develops as a terminal feature in patients with pulmonary fibrosis, it seldom, if ever, develops in the case of pulmonary fibrosis associated with HPS. In regard to the distribution of the fibrosis in HPS, it has been described as involving the respiratory bronchioles and surrounding areas, in contrast to the more peripheral alveolar localisation in usual interstitial pneumonia. Some patients with lung fibrosis associated with HPS show a tendency for the subpleural region to be spared [4] and bullous changes seem unlikely to occur around the visceral pleura.
The patient also had a bleeding diathesis, owing to the presence of abnormalities in the platelet-dense granules that regulate platelet aggregation. Primary aggregation initiated by various exogenous stimuli was preserved but the secondary wave of platelet aggregation, which depends on endogenous mediators contained in the platelet-dense granules, is diminished or absent. If bleeding is uncontrollable, platelet transfusions should be administered to correct the bleeding defect because the host platelets are normal in number and in their ability to be recruited for aggregation by the contents of the dense bodies released by the transfused platelets [3].
Footnotes
Conflict of interest: None declared.
- Received June 13, 2014.
- Accepted August 11, 2014.
- ©ERS 2014










