





Abstract
Noncommunicable diseases, including cardiovascular, metabolic and respiratory diseases, among others, are the major medical challenge of the 21st century. Most noncommunicable diseases are related to the ageing process and often co-occur in the same individual. However, it is unclear whether the index disease is somehow influencing the development of the other ones (comorbidity) or whether all of them (including the index disease) simply represent the clinical expression of pathological ageing (multimorbidity). The pathobiology of ageing, chronic obstructive pulmonary disease (COPD) and concomitant disorders is complex. A new field of research, known as systems biology if applied to model systems or network medicine if applied to human beings, has emerged over the past decade or so, to address biological complexity in a holistic, integrated way. It offers, therefore, great potential to decipher the relationship between ageing, COPD and comorbidities/multimorbidities. In this State of the Art review we present the basic concepts of systems biology, use some examples to illustrate the potential of network medicine to address complex medical problems, and review some recent publications that show how a systems-based research strategy can contribute to improve our understanding of multimorbidity and age-related respiratory diseases.
Abstract
Systems medicine can tackle the complexity of noncommunicable diseases: ageing and major chronic respiratory diseases http://ow.ly/ysZkN
Introduction
On September 19, 2011, the United Nations General Assembly declared noncommunicable diseases as the major medical challenge of the 21st century [1]. Noncommunicable diseases include a number of highly prevalent diseases, such as cardiovascular diseases, chronic respiratory diseases, cancer, diabetes, neuropsychiatric disorders and arthritis [2]. Noncommunicable diseases are now the world’s leading cause of disease burden and mortality (greater than that of infectious diseases), are increasing in prevalence (even in low- and middle-income countries) mostly in relation to the ageing of the population [3], and are extremely costly, an under-appreciated cause of poverty and a limitation for economic development [2]. Importantly, most noncommunicable diseases are related to the ageing process and often co-occur in the same individual. According to Hidalgo et al. [4], “a comorbidity relationship between two diseases exists whenever they affect the same individual substantially more than chance alone”. However, it is unclear whether the index disease is somehow influencing the development of the other ones (comorbidity) or whether all of them (including the index disease) simply represent the clinical expression of pathological ageing (multimorbidity) [5].
Chronic obstructive pulmonary disease (COPD) is a prominent noncommunicable disease because of its high prevalence, rising incidence and associated economic costs [6]. Although the inhalation of noxious particles and gases, mostly through tobacco smoking, is the main risk factor for COPD, it is also characterised by abnormal lung ageing [7] and is often associated with comorbidities/multimorbidities [8] that significantly influence the prognosis and clinical course of the patients [6]. This review addresses the following points in relation to COPD, multimorbidity and ageing. First, ageing is an extremely complex process [9, 10], characterised by progressive loss of organ function and increased vulnerability to adverse outcomes and death (i.e. frailty) [11]. Secondly, lung function, like the function of most other human organs, decreases with age [12, 13], and several respiratory diseases, like COPD, are related to pathological ageing [7]. Thirdly, COPD is often accompanied by other nonrespiratory but also chronic and age-related diseases, including cardiovascular, metabolic, osteoskeletal and neurological diseases among others. Whether these are the consequence of COPD itself (i.e. comorbidity) and/or they result from shared risk factors and molecular pathways (i.e. multimorbidity) is currently unclear [14–21], and the terms “comorbidity” and “multimorbidity” are often used indistinguishably and, potentially, erroneously. Finally, many age-related diseases, including COPD, are associated with low-grade chronic systemic inflammation, so-called “inflamm-ageing” [22, 23], and this can be an important pathogenic mechanism of multimorbidity [24]. However, the inflammatory response is also extremely complex and dynamic, and the link between multimorbidity and inflammation is not well understood [24].
Biological complexity (be it related to ageing, disease pathobiology or inflammation) has been traditionally investigated using a reductionist approach, i.e. trying to study the isolated structure and function of genes, proteins, metabolites and/or cellular organelles [25, 26]. This research strategy has been extremely successful and probably reached its summit in 2001 with the publication of the full sequence of the human genome [27]. However, there is now consensus that the building of post-genomic medicine requires an integrative approach in order to best transfer all this molecular information to clinical practice [25, 26, 28–30]. To move in that direction, a new field of research, known as systems biology if applied to model systems [31] or network medicine if applied to human beings [32, 33], has emerged over the past decade or so, to address biological complexity in a holistic, integrated way. Hence, systems biology and network medicine offer great potential to decipher the relationship between ageing, multimorbidity and lung diseases [2, 29]. In this State of the Art review we aim to present the basic concepts of systems biology, use some examples to illustrate the potential of network medicine to address complex medical problems, and review some recent publications that show how a systems-based research strategy can contribute to improving our understanding of multimorbidity and age-related respiratory diseases.
Systems biology: basic concepts
Systems biology is a novel scientific discipline that seeks to address biological complexity by integrating data within and between different levels of biological complexity (genes, molecules, cells, tissues, organs and even society and environment), modelling it and then understanding its emergent properties, i.e. those properties that cannot be explained by any of the individual components of the system [29, 32, 34, 35]. A system is not just a catalogue of genes and proteins, and its properties cannot be fully understood by simply drawing diagrams of their interconnections [34]. Systems biology tries to understand how they inter-relate and respond to external perturbations in a dynamically changing system [34]. According to Kitano, one of the founders of systems biology, this requires the understanding of four key properties of the system [34]: 1) system structures, which include the network of gene interactions and biochemical pathways, and how they modulate the physical properties of intracellular and multicellular structures; 2) system dynamics, which is how the system behaves over time and how it responds to different perturbations; 3) control methods, used to minimise malfunction of the systems, which, interestingly, can become potential therapeutic targets for treatment of disease; and 4) design methods, which can be used to modify and/or construct biological systems with specifically desired properties, instead of blind trial and error.
To achieve these goals, systems biology generally follows an iterative sequential strategy. This involves the use of high-throughput platforms to collect genome, transcriptome, proteome and metabolome information (collectively known as “omics” techniques), sometimes complemented with information existing in publicly available databases through knowledge management platforms [36]. Bio-computing algorithms are used to generate multiscale (from the molecular to the organ level) predictive mathematical models [37], then these models are used to formulate novel working hypotheses on the mechanisms and pathways involved in the disease of interest. These novel hypotheses are tested through perturbation experiments, which can be done in silico (computer model simulation), in vitro (cell culture), or in vivo (animal models), then the experimental responses observed are compared with those predicted by the initial mathematical models, which are then refined accordingly. This process is iterated until the derived model predicts, with reasonable accuracy, the observed experimental findings, at which point the model allows a better understanding of the disease of interest and the identification of novel biomarkers [38]. Clearly, therefore, systems biology requires multidisciplinary collaboration, as does network medicine, as will be described in the next section.
Network medicine: some examples
Human health and disease are emergent properties of an extremely complex system (the human body) that relies on the interaction of many components, within and across cells and organs [29, 30]. The potential complexity of this network, the so-called “human interactome”, is daunting: ∼25 000 protein-coding genes, ∼1000 metabolites, an undefined number of distinct proteins and functional RNA molecules, and more than 100 000 cellular components, which all serve as the nodes of the human interactome [33]. This intra- and intercellular connectivity implies that the impact of a specific abnormality is not restricted to the activity of that individual component of the interactome, but can spread along the links (connections or edges) of the network and alter the activity of other components, hence modulating the final phenotypic expression of the original molecular abnormality [33]. In essence, the basic hypothesis of network medicine is that human diseases arise as a consequence of one or more biological networks in the relevant organ that have become disease-perturbed through genetic and/or environmental pathogenic changes [25].
This is illustrated in figure 1, which shows that, contrary to the original concept of a “gene” as the “determinant of the phenotype” (vertical arrow), the current notion of a “gene” is that it is merely a DNA sequence whose influence on the phenotype is modulated by environmental influences through biological networks (circle), which include signalling pathways, conditioners, buffers and incubators that enable and restrict reactions [39]. This network perspective has direct implications for the understanding of the relationship between age-related multimorbidity and respiratory diseases, since a disease phenotype in complex diseases is rarely the consequence of an abnormality in a single effector gene product, but reflects various pathobiological processes that interact in this complex network [33], and there may be functional, molecular and causal relationships among apparently distinct phenotypes [33].

Relationships between genes, environment and phenotypes. Genes were originally conceived as the “determinants of a phenotype” (vertical arrow). However, the current biological notion of a gene is that of DNA sequences whose phenotypic expression depends on complex interactions with the environment mediated by biological networks, which enable, filter, condition and buffer them. Reproduced and modified from [39] with permission from the publisher.
Following these realisations, Barabási, one of the founders of network medicine [33], proposed the term “diseasome” to illustrate that, although often treated separately, most human diseases are not independent of each other [32]. As shown in figure 2, one way to visualise the inter-relationships among human diseases is to construct a disease network (middle layer), in which two diseases are connected if they have a common genetic, regulatory or metabolic origin and/or common protein–protein interactions (bottom layer) [32]. The social–environmental network (top layer) is also of key importance to human disease (fig. 3). In the case of respiratory diseases, this top layer includes smoking and exposure to other toxic environmental gases and particles, infections and allergens, which may interact (cold temperatures interact with viral infections, spring with allergens, etc.) and form a true network themselves too [40, 41]. In other chronic diseases, such as obesity, human-to-human interactions have been shown to have a key pathogenic role [42].

Graphical illustration of the diseasome (middle layer), where two diseases are connected if they have a common genetic or functional origin (bottom layer). A third relevant network for human disease is the social network (top layer), which encompasses all human-to-human interactions and may play a role in the spread of infectious and noninfectious (obesity, smoking) diseases. Reproduced from [32] with permission from the publisher.

Diagram illustrating the different levels of complexity of chronic obstructive pulmonary disease (COPD) and the outcomes of potential clinical relevance (right-hand column). CVD: cardiovascular disease; GWAS: genome-wide association studies; miDNA: mitochondrial DNA; miRNA: microRNA; ncRNA: noncoding RNA. Reproduced from [41] with permission from the publisher.
Barabási and co-workers also published the first description of the human diseasome (which they called the “human disease network” [43]), a network of human diseases linked by known shared susceptibility genes (fig. 4). This analysis provided several observations of great interest.
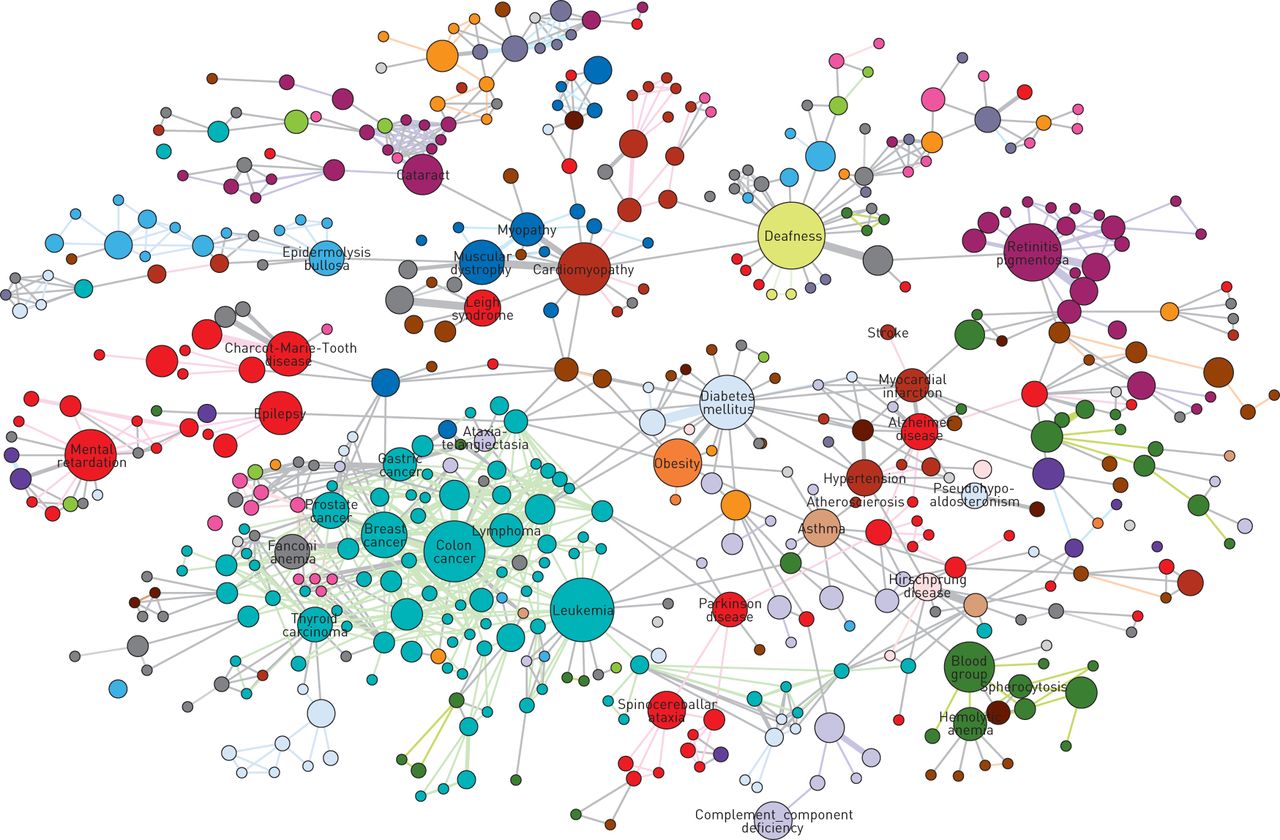
Graphical representation of the human disease network, where each node corresponds to a distinct disorder and colours represent disease classes. The size of each node is proportional to the number of genes participating in the corresponding disorder, and the thickness of the edge (link) is proportional to the number of genes shared by the disorders it connects. Reproduced and modified from [43] with permission from the publisher.
First, like many other biological networks, the human disease network has a scale-free structure. As shown in figure 5, scale-free networks are a particular type of network characterised by having the majority of the nodes (i.e. the core elements of the network, whether they are genes, proteins, metabolites or whatever) connected to other nodes of the network by a relatively small number of links, whereas a few nodes (hubs) present an extremely high number of links [44, 45]. This is very different from the Poisson network, where the majority of nodes have a similar (and relatively small) number of links. Scale-free networks are differentially sensitive to damage [46]. This means that if a peripheral node stops functioning, the network is very likely to continue working without problem. By contrast, if a hub is damaged, the functionality of the entire network is likely to be jeopardised [46].
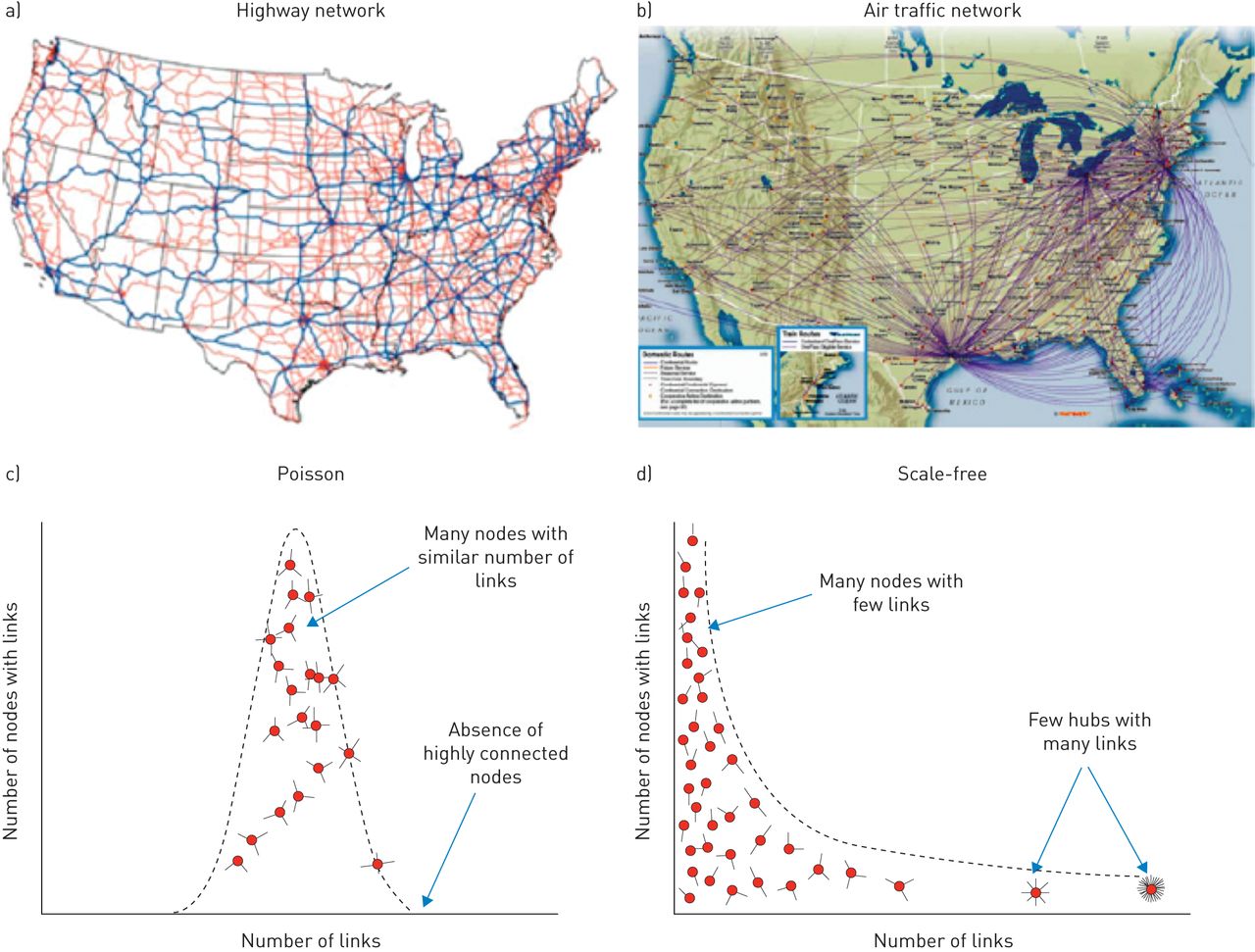
Poisson and scale-free networks. a) The highway network in the USA is an example of c) a Poisson network: most cities have a relatively small and similar number of highways interconnecting them. By contrast, b) the airport network in the USA is a good example of d) a scale-free network: only a few airports (hubs) host a large number of flight connections, whereas the majority of airports have only a few connections. Reproduced and modified from [29] with permission from the publisher.
Secondly, whereas the vast majority of disease genes were nonessential, showed no tendency to encode hub proteins and their expression pattern indicated that they were localised in the functional periphery of the network [43], essential genes encoded hub proteins that were expressed widely in most tissues [29].
Finally, genes associated with similar diseases exhibited a higher likelihood of physical interaction of their products and a higher expression profiling similarity of their transcripts, supporting the existence of shared “disease modules” [43]. Disease modules are a “group of network components that together contribute to a cellular function and disruption of which results in a particular disease phenotype” [33]. Disease modules can be identified by combining high-throughput “omic” results in carefully designed experiments and bioinformatic analysis using the interactome of all known protein–protein interactions, DNA–protein interactions and human metabolic pathways [33].
It is important to note that the interactome is currently incomplete [33]. In any case, the human disease network illustrates the potential of network medicine to systematically explore both the molecular complexity of a particular disease, leading to the identification of disease modules and pathways that may become targets for drug development, and the molecular relationships among apparently distinct (patho) phenotypes (multimorbidity). Furthermore, network medicine can facilitate the identification of better and more accurate biomarkers, to monitor the functional integrity of networks that are perturbed by diseases, and can lead to novel and better disease classification taxonomies [30, 33]. Rzhetsky et al. [47] published similar results using 161 disorders in 1.5 million patient records.
A network approach to ageing, multimorbidity and respiratory diseases
The three processes considered here (ageing, multimorbidity and chronic respiratory diseases) are, each of them, extremely complex. There have been a few attempts to address their complexity using systems biology and network medicine approaches. These are reviewed below.
Network approaches to ageing
Ageing is a complex process [9, 10]. López-OtÍn et al. [10] have recently proposed nine hallmarks of ageing (fig. 6): genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication. A major scientific challenge is to understand their relationships and relative contribution to normal and pathological ageing (fig. 6) [10].
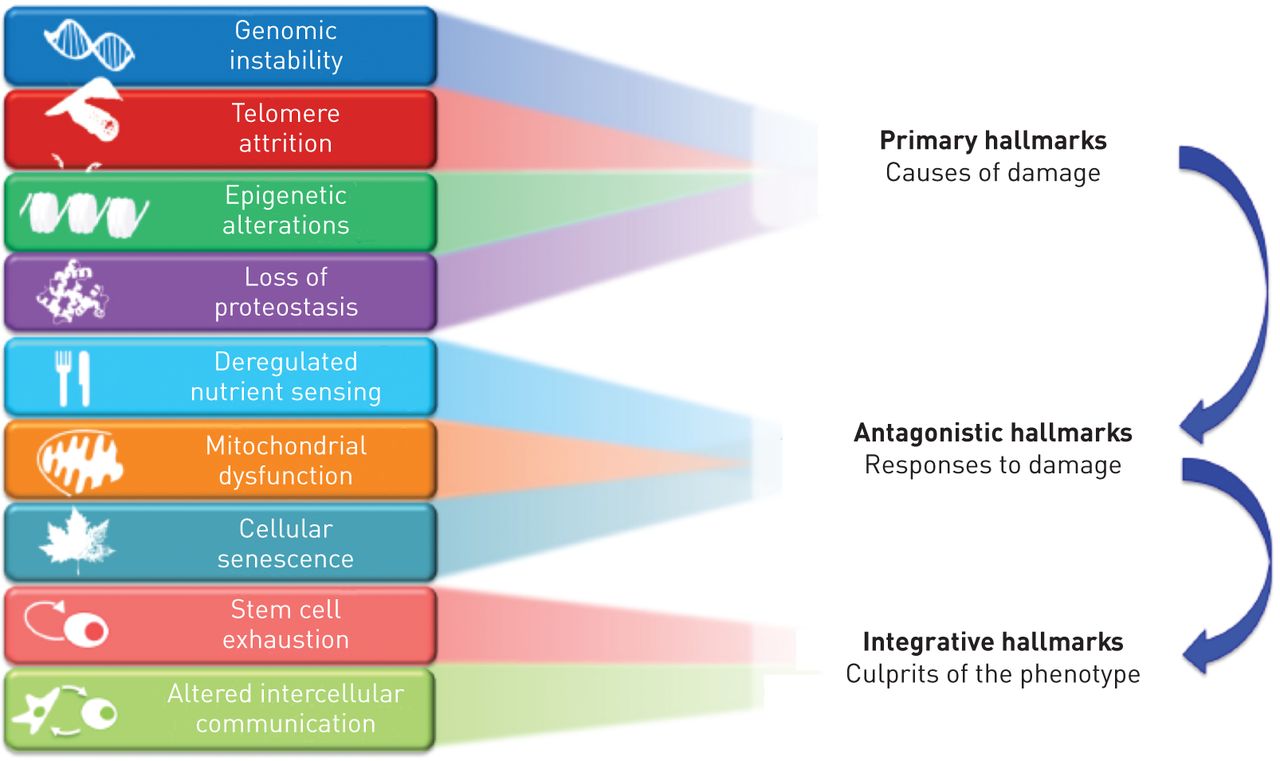
Hallmarks of ageing categorised into primary causes of cellular damage (top), compensatory or antagonistic responses (middle), and those ultimately responsible for the functional decline associated with ageing (bottom). Compensatory responses initially mitigate damage, but eventually, if chronic or exacerbated, they may become deleterious themselves. Reproduced from [10] with permission from the publisher.
Several studies have already used a systems biology/network medicine approach to study the complexity of ageing [48]. For instance, Sozou et al. [49] investigated the interactions of the different factors known to influence cell sencescene, including oxidative damage, telomere shortening and mitocondrial and nuclear DNA mutations, whereas other groups studied the relationships between dietary restriction, metabolic regulation and longevity in model organisms [50–52]. In humans, the study of centenarians led to the identification of a number of loci associated with longevity [53]. Likewise, Xue et al. [54] used age-dependent gene expression data and protein–protein interactions (i.e. the interactome) of transcriptionally correlated genes to create an active ageing sub-network, where hubs were enriched in lifespan regulator genes [54]. Interestingly, the in silico removal of these genes destabilised the network structure, suggesting that ageing preferentially targets regulators controlling the organisation and coordination of temporal switches [54, 55]. Although much more work remains to be done in order to understand the complexity of normal ageing and to extrapolate this new knowledge to pathological ageing and disease, it is clear that studies using network-based approaches like those briefly reviewed here have just begun to unravel them.
Network approaches to multimorbidity
As discussed, the diseasome is the network representation of human disease (nodes of the network) linked by shared disease-associated cellular components [4]. To date, three types of disease-linking networks, based on three different formalisms, have been described: 1) gene formalism, where diseases are linked based on shared genes [43]; 2) metabolic formalism, where connections reflect shared metabolic pathways [56]; and 3) disease comorbidity formalism, where links between diseases are based on their co-occurrence in excess to what is expected by chance [4]. The following three studies used these formalisms to understand the complex network structure of comorbidity.
Lee et al. [56] used the gene and metabolic formalisms to explore the relationship among different diseases, which were linked if mutated enzymes associated with them were responsible for catalysing adjacent metabolic reactions. They found that connected disease pairs display higher correlated reaction flux rate, corresponding enzyme-encoding gene co-expression, and higher comorbidity than those that have no metabolic link between them [56]. Furthermore, the more connected a disease was to others, the higher was its prevalence and associated mortality rate [56].
Park et al. [57] used the disease comorbidity formalism to investigate the presence of significant correlations between diseases in the Medicare registry, including more than 13 million individuals. To this end, they also used the shared genes formalism and combined information on omics, shared genes, protein–protein interactions and co-expression patterns, and found statistically significant correlations between cellular interactions and comorbidity patterns, and that disease pairs with higher correlations tend to be linked more strongly at the cellular network level [57]. These results not only provide relevant biological information but also suggest that Medicare and other insurance databases could play an increasing role in future studies of the systems biology of human cells and diseases [57].
Finally, Hidalgo et al. [4] published the so-called “phenotypic disease network”, which summarises comorbidity correlations obtained from the disease history in the Medicare registry, and, importantly, investigated the dynamics over time of the phenotypic disease network. The main results showed that patients develop diseases close in the network to those they already have, albeit with apparent sex and ethnic differences, and that patients diagnosed with diseases that are more highly connected in the phenotypic disease network tend to die sooner than those affected by less connected diseases [4]. Overall, these observations indicate that disease progression can be represented and studied using network analysis [4].
Network approaches to chronic respiratory diseases
Some recent studies have used network approaches to study the pathobiology of several chronic respiratory diseases, including asthma [58], COPD [24, 59] and idiopathic pulmonary fibrosis [60]. Due to its frequent relationship with multimorbidity, we will focus here on those addressing COPD.
The Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) study was a large 3-year observational controlled multicentre international study (NCT00292552) aimed at defining clinically relevant subtypes of COPD and identifying novel biomarkers and genetic factors [61]. The results of the ECLIPSE study have significantly influenced our understanding of COPD [62, 63]. Using data from ECLIPSE, AgustÍ et al. [24] described the so-called “systemic inflammome” of smoking and COPD, a network representation of systemic inflammation in these individuals. Importantly, this network analysis showed that: 1) the inflammome of smoking and COPD are different; 2) 30% of COPD patients are never inflamed whereas about 20% of them are persistently inflamed; and 3) despite similar lung structural and functional abnormalities, the latter had a six times higher all-cause mortality (and double the incidence of exacerbations) than the former during 3 years’ follow-up [24], hence identifying a novel phenotype that requires research and, potentially, specific treatment [64, 65].
More recently, to explore the genetic expression basis of COPD, Menche et al. [59] used the clinical (age, body mass index (BMI), tobacco smoking exposure, history of exacerbations), lung function (forced expiratory volume in 1 s, expressed as % predicted), imaging (presence/severity of emphysema and/or airway disease), systemic inflammation (high-sensitivity C-reactive protein, interleukin (IL)-6, IL-8, chemokine C-C motif ligand (CCL)18, fibrinogen, tumour necrosis factor-α, surfactant protein (SP)-D) and sputum transcriptomic (Affymetrix microarrays) data obtained in a subset of 140 patients who were also recruited into the ECLIPSE study. The authors used a dual research strategy. First, they compared the number of differentially expressed genes in the two extreme quartiles of several clinically relevant phenotypes identified a priori by COPD experts, including the presence of chronic bronchitis (i.e. regular cough and sputum production), previous history of COPD exacerbations, low/high BMI, severity of airflow limitation, presence/severity of emphysema and airway disease on computed tomography scan. Secondly, they used an alternative but complementary approach, where they identified groups of patients with maximally different gene expression patterns, and then compared their phenotypic clinical expression. To identify these groups with extremely different transcriptomic signatures, the authors developed a novel unbiased bioinformatics algorithm (the diVIsive Shuffling Approach (VIStA)), which, through an iterative approach, ended up maximising gene expression differences between groups [59]. In brief, as shown in figure 7a, the VIStA method is as follows. 1) The algorithm first randomly partitions the available patients (n=140 in this case) into three groups of comparable size, compares gene expression of groups 1 and 2 and keeps group 3 as a reservoir. 2) It randomly swaps (shuffles) one patient from group 1 (or 2) with another one from the reservoir (group 3), and differential gene expression is compared again. If the new number of differentially expressed genes increases, the swap is accepted, if not it is rejected. 3) Step 2 is iterated until the number of differentially expressed genes reaches a plateau, generally after approximately 1000 attempted swaps. At this point, the resulting groups 1 and 2 include patients with the maximal gene expression differences [59].

a) Schematic representation of the diVIsive Shuffling Approach (VIStA). b) Network representation of the clinical characteristics identified by VIStA. Node size is proportional to the number of times a given clinical characteristic was found to be significant in VIStA, and the width of a link indicates how often two measures appeared significant in the same VIStA division. The core group (hub) contains severity of airflow limitation (GOLDCD) and the qualitative (EMPHETCD) and quantitative (FV950) measures of emphysema. SPD: surfactant protein D; TNFA: tumour necrosis factor-α; FIBRINOG: fibrinogen; CCL18: chemokine C-C motif ligand 18; DWALK: 6-min walking distance; CRPHS: high-sensitivity C-reactive protein; IL: interleukin; BMI: body mass index. Reproduced and modified from [59] with permission from the publisher.
The results of the first approach, i.e. the comparison of differentially expressed genes in the extreme quartiles of the clinical phenotypes defined a priori by COPD experts, failed to identify any difference, except for the severity of airflow limitation, where 6049 differentially expressed genes were identified [59], indicating that mild/moderate and severe/very severe COPD are associated with significantly different transcriptomic signatures in sputum. The results of the reverse approach using VIStA showed that, after about 500 VIStA runs, the severity of airflow limitation, this time in combination with the amount and severity of emphysema present, was again the most important hub of the network (fig. 7b) [59]. Interestingly, however, the investigators also observed that age, BMI, exercise capacity, chronic bronchitis, some inflammatory biomarkers (IL-6, IL-8 and SP-D) and some sputum findings (high number of neutrophils and low number of lymphocytes) provide further discriminant power [59]. In summary, the results of the study by Menche et al. [59] illustrate the potential of systems biology and network medicine to address the complexity of chronic respiratory diseases like COPD. Specifically, they demonstrate that mild/moderate versus severe/very severe COPD are associated with different sputum transcriptomic signatures. What is cause and effect cannot be dissected from this cross-sectional analysis, but these observations are certainly compatible with the hypothesis that mild/moderate and severe/very severe COPD might be two different diseases.
Putting ageing, multimorbidity and COPD together using systems/network approaches
Several articles have attempted to put together multimorbidity, age and COPD. Divo et al. [19] evaluated the prevalence of comorbidities in patients with COPD attending a pulmonary clinic and assessed their relationship with mortality (fig. 8). They observed that 12 specific comorbidities were significantly associated with an increased risk of death. They represented graphically the prevalence of these comorbidities and their relationship with the risk of death as the comorbidome [19].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The “comorbidome” is a graphic expression of comorbidities with more than 10% prevalence in the entire cohort, and those comorbidities with the strongest association with mortality. The area of the circle relates to the prevalence of the disease. The proximity to the centre (mortality) expresses the strength of the association between the disease and risk of death. All circles associated with a statistically significant increase in mortality are fully inside the dotted orbit. OSA: obstructive sleep apnea; CVA: cerebrovascular accident; HTN/RHF: hypertension and right heart failure; CHF: congestive heart failure; PAD: peripheral artery disease; CAD: coronary artery disease; A. fibrillation: atrial fibrillation/flutter; BPH: benign prostatic hypertrophy; CRF: chronic renal failure; DJD: degenerative joint disease; GORD: gastro-oesophageal reflux disease. Reproduced and modified from [19] with permission from the publisher.
Vanfleteren et al. [66] identified five clusters of patients based on 13 objectively identified comorbidities and measured systemic inflammation in 213 COPD patients attending a rehabilitation programme. The five clusters were: “less comorbidity”, “cardiovascular”, “cachectic”, “metabolic” and “psychological”. These clusters differed in health status but were comparable in terms of airflow limitation severity and systemic inflammatory markers [66]. Unfortunately, no longitudinal data or relationships with clinically relevant outcomes were provided.
Van Remoortel et al. [21] have recently investigated the effects of smoking and ageing on the prevalence of premorbid risk factors and comorbid diseases, as well as their association with daily physical activity (by multisensor activity monitor), in the preclinical stages of COPD. They studied a population-based sample of subjects (n=60) with a new diagnosis of mild-to-moderate COPD by spirometry screening. The results were compared with those of 60 smoking controls with normal lung function and 60 never-smoker subjects, all of them age matched [21]. The main results showed that premorbid risk factors and comorbid diseases were significantly higher in preclinical COPD than in never-smokers but similar to smoking controls; cardiovascular diseases and musculoskeletal dysfunction were particularly prevalent in smokers with or without COPD, and other diseases that were more frequently found in the smoking group included pre-diabetes, systemic arterial hypertension, dyslipidaemia and obesity. Multivariate logistic regression analysis showed that physical inactivity and smoking were independent risk factors for suffering two or more comorbidities [21]. Overall, these observations challenge the concept that COPD is an independent risk factor for comorbidities by showing that physical inactivity and smoking, but not COPD as such, are associated with their development [21]. Overall, these results are in keeping with other observations in COPD patients, showing that the prevalence of comorbidities is independent of the severity of airflow limitation (Global Initiative for Chronic Obstructive Lung Disease (GOLD) grades) [66–68], and that age rather than COPD severity accounted for most of the comorbidities of these patients [69].
Other interesting observations that used a systems biology approach in COPD include: the study by Xie et al. [70], who identified the serum levels of the microRNAs miR-21 and miR-181a as potential biomarkers of COPD susceptibility among heavy smokers; that of Ezzie et al. [71], who identified 70 microRNAs and 2667 mRNAs differentially expressed in lung tissue from smokers with and without COPD [71]; and that of Turan et al. [72], who investigated the relationship between skeletal muscle dysfunction, a frequent and clinically relevant comorbidity in COPD [73, 74], pulmonary gas exchange, systemic inflammation and response to training. The results of Turan et al. [72] indicate that, in COPD, skeletal muscle fails to co-ordinately activate the expression of several remodelling and bioenergetics pathways, and this may be linked to an abnormal expression of histone modifiers, which, in turn, appears to correlate with tissue oxygen utilisation. Overall, these observations suggest that cell hypoxia may be a key factor driving skeletal muscle dysfunction in COPD patients [72]. This contrasts with current hypotheses based on the role of systemic inflammation and suggests an alternative oxygen-driven, epigenetic control mechanism [72].
Finally, AgustÍ et al. [75] have recently proposed a network approach to the pathobiology of COPD at the organ and molecular levels. The basic tenant of this approach is that COPD is not a single-organ condition, and that disturbances of a complex network of inter-organ connected responses occur and modulate the natural history of the disease [75]. The acceptance of such inter-organ connectivity leads to the appreciation that toxic inhaled agents that directly affect the lungs are also likely to exert effects (direct or indirect) on more distant organs. These, in turn, might modulate the lung’s own response (be it acute or chronic) to the initiating injury [75]. Specifically, the authors propose a vascularly connected network where the lungs are the main external sensor of the system and a source of so-called “danger signals”, the endothelium acts as an internal sensor of the system (and also is a potential target tissue) and the bone marrow and adipose tissue are two key responding elements that produce both inflammatory and repair signals. According to this novel network model, the development of COPD and associated multimorbidities would depend on how this vascular connected network responds, adapts or fails to adapt (dictated by the genetic and epigenetic background of the individual) to the inhalation of particles and gases, mainly in cigarette smoke [75].
Conclusions
Many noncommunicable diseases, including chronic respiratory diseases like COPD, are associated with ageing and are often accompanied by other noncommunicable diseases (multimorbidity). The pathogenesis of each of them (including that of the ageing process), as well as their inter-relationships at the molecular, clinical and environmental levels, are extremely complex and dynamic. This State of the Art review highlights that systems biology and network medicine offer a new research strategy to decipher this multilevel and dynamic complexity, by reviewing some recent investigations in the field. We firmly believe that this is only the beginning of a new way of understanding and, eventually, diagnosing and treating what, as stated before, has been identified by the United Nations General Assembly as the major health challenge at the beginning of the 21st century: the growing epidemic of age-related noncommunicable diseases. Stay tuned!
Footnotes
Support statement: This work was supported, in part, by FIS 12/01117, SEPAR 192/2012, BECA FUCAP 2013, BECA SEPAR 065/2013, RESERCAIXA 2012, 2014FIB00417 and BFI-2012-66.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received April 28, 2014.
- Accepted June 16, 2014.
- ©ERS 2014
References










