





Abstract
Glutathione is an important antioxidant in the lungs but its concentration is low in the airways of patients with cystic fibrosis. Whether this deficit occurs from an early age or how oxidative stress contributes to lowering glutathione is unknown.
We measured glutathione, its oxidation products, myeloperoxidase, and biomarkers of hypochlorous acid in bronchoalveolar lavage from children with cystic fibrosis and disease controls using mass spectrometry and immunological techniques.
The concentration of glutathione was lower in bronchoalveolar lavage from children with cystic fibrosis, whereas glutathione sulfonamide, a specific oxidation product of hypochlorous acid, was higher. Oxidised glutathione and glutathione sulfonamide correlated with myeloperoxidase and a biomarker of hypochlorous acid. The percentage of glutathione attached to proteins was higher in children with cystic fibrosis than controls. Pulmonary infections in cystic fibrosis resulted in lower levels of glutathione but higher levels of oxidised glutathione and glutathione sulfonamide in bronchoalveolar lavage.
The concentration of glutathione is low in the airways of patients with cystic fibrosis from an early age. Increased oxidation of glutathione by hypochlorous acid and its attachment to proteins contribute to this deficiency. Therapies targeted against myeloperoxidase may boost antioxidant defence and slow the onset and progression of lung disease in cystic fibrosis.
Abstract
The antioxidant glutathione is low in the airways of children with CF due to oxidation by hypochlorous acid http://ow.ly/tKfyX
Introduction
Cystic fibrosis (CF) is the most common fatal hereditary disease in Caucasians [1]. It is caused by mutations in the CF transmembrane conductance regulator (CFTR) protein that alters the transport of small negatively charged ions across the lung epithelia. These anions include chloride and bicarbonate. Numerous theories have been proposed to link the defect in the CFTR to lung disease [2]. The current thinking is that a lack of bicarbonate leads to a decrease in pH [3], as well as improper secretion and swelling of mucins [4]. Ultimately, all the theories propose to an imbalance of anion transport in the CF airways, which makes the lungs susceptible to repeated bouts of bacterial infection and inflammation. Progressive lung disease then develops and results in considerable morbidity and premature death [2]. The predominant inflammatory cells that infiltrate the epithelium are neutrophils [5]. These phagocytic cells attempt to trap and kill bacteria but in their frustrated attempts to do so, they release proteases and reactive oxygen species that damage the airways [6].
Glutathione (GSH) is also transported by the CFTR [7] and acts as an antioxidant in the epithelial lining fluid (ELF) [8], [9]. The concentration of this anionic tripeptide (γ-Glu-Cys-Gly) in the ELF of normal individuals is ∼400 μM, which is over a 100-fold higher than in plasma [10]. Analysis of bronchoalveolar lavage (BAL) from young adults with CF indicated that the concentration of GSH is low in their ELF and should compromise antioxidant defence [11]. In a similar analysis of infants and young children with CF, GSH tended to be lower in those with an infection when compared to the controls, but the difference was not found to be significant [12].
GSH acts as an antioxidant in ELF by scavenging reactive oxygen species [13]. In the inflamed CF airways, neutrophils are the major source of oxidants [14]. These phagocytes generate superoxide and hydrogen peroxide, and use the enzyme myeloperoxidase (MPO) to convert hydrogen peroxide to an array of reactive oxygen species; including hypochlorous acid, and radicals such as those from urate and tyrosine [15]. GSH reacts readily with most of these oxidants [16]. Consequently, in CF the antioxidant shield provided by GSH could be overwhelmed. Proteins in BAL from children with CF are highly chlorinated, suggesting that considerable hypochlorous acid is produced during inflammation and GSH concentrations are inadequate to stop it from damaging the airways [14], [17].
When hypochlorous acid oxidises GSH it produces predominantly oxidised GSH (GSSG) and forms GSH sulfonamide (GSA) as a minor, but specific, product [18]. In the presence of proteins, there is potential for GSH to form mixed disulfides with cysteine residues in a process known as S-glutathionylation [19].
We undertook this study to establish whether the concentration of GSH is low in the ELF of young children with CF and test the hypothesis that this arises from oxidation by activated neutrophils.
Materials and methods
Characterisation of CF patients
This retrospective analysis included 167 subjects, born between 1993 and 2009, diagnosed with CF and enrolled in the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) Early Surveillance Program (ESP) in Perth and Melbourne, Australia [20], [21]. Further details of the programme are provided in the online supplementary material. All aspects of the ESP have been approved by the human research ethics committees at each centre. Parents provided written consent at each assessment. Clinic participation rates for the AREST CF ESP were over 90%, with 85% of the children in the study undergoing annual computed tomography (CT) and bronchoscopy with BAL for the duration of the ESP.
The 21 control subjects were children without CF (non-CF) who underwent BAL for assessment of persistent respiratory symptoms, including stridor, chronic cough, or recurrent pneumonia.
Chest CT
Chest CT scans were performed as previously described [21]. In brief, children were anaesthetised and intubated using a cuffed tube, with a recruitment manoeuvre to reduce procedure related atelectasis. A volume-controlled limited slice scan was performed with three slices taken at end-inspiration (25 cmH2O) and end-expiration (0 cmH2O) as previously described, with lung volume controlled by the anaesthetist, using a Philips Brilliance 64 (Philips Medical Systems, Eindhoven, the Netherlands) CT scanner in Perth and a Somatron Sensation 16 (Siemens Medical Solutions, Erlangen, Germany) CT scanner in Melbourne, Australia.
Images were scored in batches of 5–20 as they became available by a single scorer as previously published on a soft copy reporting station (Agfa Impax TM, Agfa-Gevaert, Mortsel, Belgium) using standard lung window protocols. While the scorer was aware of the diagnosis of CF, scans were assessed without knowledge of the outcome of any previous scans, clinical status, microbiology or inflammatory results. Each scan was divided into six zones (left and right upper, middle and lower zones), corresponding with each axial slice. Each zone was scored for the presence of bronchiectasis and air trapping using a previously reported modification of a CF specific CT scoring system [21], [22]. Bronchiectasis was scored on inspiratory images, defined as a bronchus-to-artery ratio >1, or nontapering bronchus visible in transverse plane [20]–[22].
BAL
BAL was performed under general anaesthesia following the CT scan. Three aliquots of normal saline, each with a volume of 1 mL·kg−1, were instilled into the right middle lobe or right lower lobe and retrieved using low-pressure suction. The first aliquot was used for microbiological analysis. The remaining aliquots were pooled, centrifuged at 167×g for 5 min and the supernatant was decanted, aliquoted, and frozen at -80°C for future biochemical analysis.
The cell pellet from the pooled samples was used for cytology, and total cell counts were standardised for the volume retrieved in those pooled samples. One aliquot of BAL was split in two and 10 mM N-ethylmaleimide (NEM) was added to one of the samples to stabilise GSH by alkylation. Due to volume constraints, not all samples were tested for all of the analytes studied. To minimise oxidation during storage, samples were kept at -80°C and thawed only a maximum of two times. Also, the addition of NEM to GSH and protein thiols samples protects from artefactual oxidation.
BAL-detected infection
BAL samples were considered “infected” with bacterial, viral or fungi if there was a positive culture for at least one dominant organism at any density of ≥105 CFU·mL−1 or with Pseudomonas aeruginosa at any density [21]. The culture of mixed oral flora, without a predominant organism was considered to be “uninfected”.
BAL-detected inflammatory analyses
The second and third BAL aliquots from the right middle lobe were pooled and processed for inflammatory analyses as previously described including total and differential cell count, interleukin (IL)8 concentrations, using ELISA, and the detection of free neutrophil elastase (NE) activity [17], [21]. The lower limit of detection for IL-8 and NE were 100 pg·mL−1 and 200 ng·mL−1, respectively.
Biochemical analysis
Total protein was measured using the Bio-Rad detergent-compatible assay kit (Bio-Rad Laboratories Inc Hercules, CA, USA). MPO activity and protein were measured by an activity ELISA [23]. BAL protein was hydrolysed and analysed for tyrosine and chlorinated tyrosine by gas chromatography-mass spectrometry with isotopically labelled internal standards [17]. Results are expressed as 3-chlorotyrosine mmol residues per mol tyrosine residues (mmol:mol) and were corrected for artifactual tyrosine chlorination (typically≤10 μmol:mol).
Analysis of GSH and its oxidation products in BAL
GSH, GSSG and GSA was analysed by stable isotope dilution liquid chromatography tandem mass spectrometry assay (LC-MS/MS) as previously described [24]. Aliquots of standards (0·2 mL) or BAL (a volume containing 50 μg total protein made up to 0·2 mL) were mixed with isotopically labelled GSH, GSSG and GSA as internal standards. Ice-cold ethanol (0·8 mL) was added and precipitated protein pelleted. Supernatants were aspirated, and dried under vacuum. Residues were redissolved in 200 μL of phosphate-buffered saline, and 50 μL was injected onto the column.
S-glutathionylation of proteins in BAL was detected by precipitating proteins with ice-cold ethanol, washing the precipitate once and then adding 10 mM dithiothreitol to liberate the GSH from the proteins. The GSH, in these samples, was alkylated with 10 mM NEM and then measured by using stable isotope-dilution mass spectrometry as described earlier. The CF samples that were analysed were selected from the sample data base using a random number generator.
Statistical analysis
Results are expressed as mean±sd of untransformed data, unless otherwise stated. Non-normally distributed data were log (ln) transformed for statistical analyses. Analyses for comparisons between groups were completed using t-tests or a regression model for addition of confounding variables. Where appropriate, age was added as a confounding variable. Associations between age, inflammatory markers and analytes were compared using a Pearson correlation using log (ln) transformed data. A p-value of <0·05 was considered significant. The mean of the measurements was used when an analyte was measured more than once in a BAL sample. In the cases where an analyte was below the limit of detection in a BAL sample, a value of half the lower limit of detection was assigned for statistical purposes. Statistics were performed using STATA (Version 11.0, StataCorp LP, College Station, TX, USA), SigmaPlot and SigmaStat software (Jandel Scientific, San Rafael, CA, USA).
Results
GSH concentrations are low in the airways of children with CF
Children with CF were slightly younger than the non-CF disease controls, and had elevated inflammatory cell counts, including macrophages and neutrophils, elevated IL-8, NE and MPO, and higher levels of 3-chlorotyrosine present in the proteins from BAL (table 1).
The concentration of GSH in BAL from children with CF was approximately half of that measured in the disease controls (table 1 and fig. 1). There was no significant difference in the concentrations of GSSG (table 1) or the proportion of GSH present as GSSG. For the majority of children with or without CF, GSH was present in a large excess over GSSG (table 1).
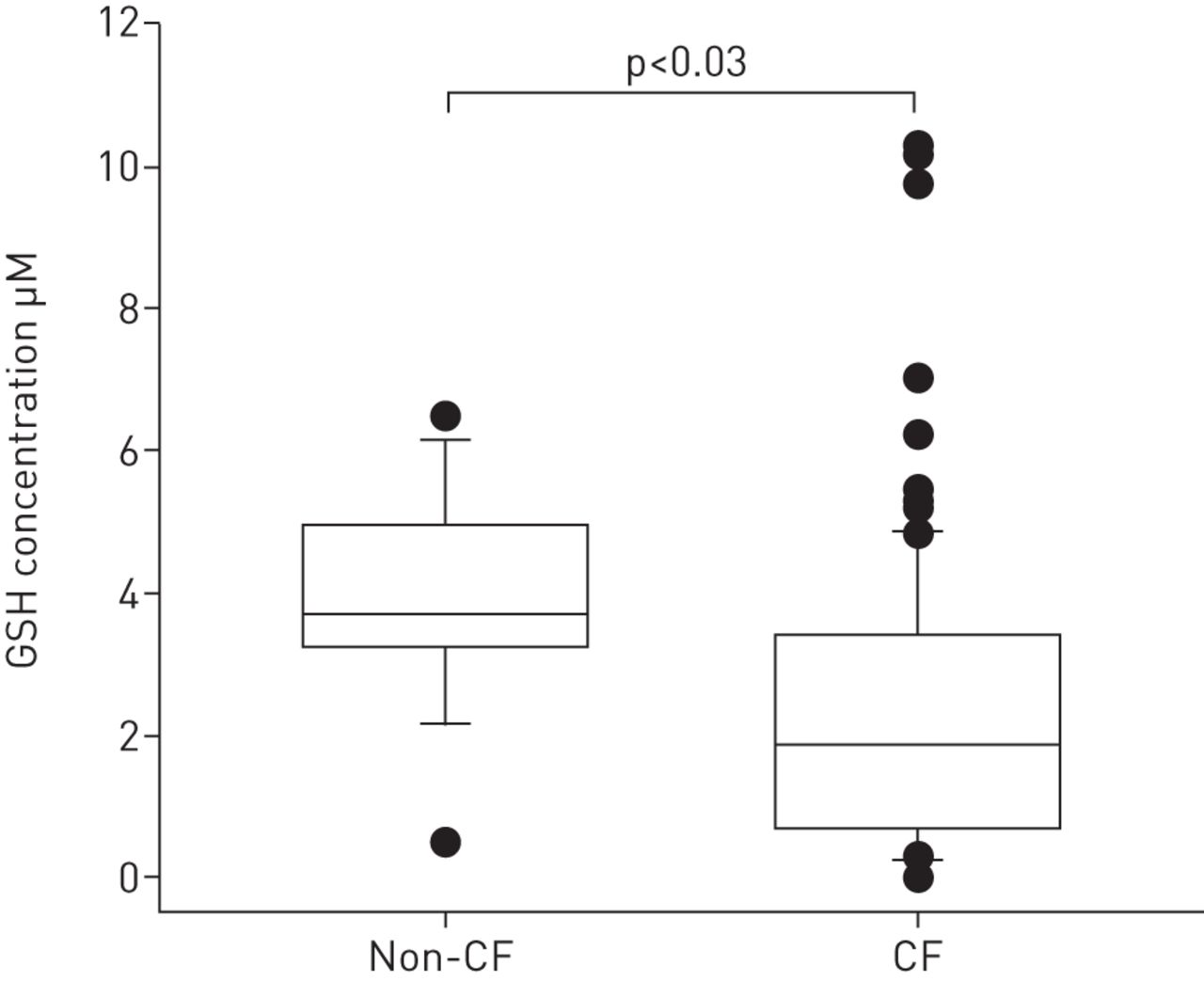
Glutathione (GSH) bronchoalveolar lavage (BAL) from children with cystic fibrosis (CF) and without CF (non-CF). GSH was measured in BAL samples taken from CF children (n=167) and non-CF children (n=21) using liquid chromatography with stable isotope dilution mass spectrometry. Data are presented as the absolute concentration of GSH. Differences between the groups were calculated on log (ln) transformed data using a regression analysis with age added as a confounding variable. The lower boundary of the boxes represents the 25th percentile, the line within the boxes is the median, and the upper boundary represents the 75th percentile. The whiskers represent the 10th and 90th percentiles while filled circles are values that lie outside these percentiles.
Hypochlorous acid oxidises GSH in the airways
GSA was present in BAL and its absolute concentration tended to be higher in CF BAL and accounted for a significantly higher percentage of total free GSH (table 1 and fig. 2a). Most children had <1% of the total GSH present as GSA although some had up to 5% or more present as this form. GSA was strongly correlated with 3-chlorotyrosine (r=0·72, p<0·0001), a specific biomarker of hypochlorous acid (fig. 2b and table E1) and MPO (r=0·57, p<0.0001) (table E1). It was also strongly correlated with markers of inflammation including neutrophil density, neutrophil elastase and IL-8 in children with and without CF (tables E1 and E2).

Glutathione sulfonamide (GSA) in bronchoalveolar lavage (BAL) from children with cystic fibrosis (CF) and without CF (non-CF). a) GSA was measured in BAL samples from CF children (n=167) and non-CF children (n=21) using liquid chromatography with stable isotope dilution mass spectrometry. Data are presented as the absolute concentration of GSA. b) The relationship between GSA and the content of 3-chlorotyrosine of proteins in BAL from children with CF (n=70). Data were analysed using a Pearson correlation on log (ln) transformed data.
Oxidation of GSH is associated with biomarkers of hypochlorous acid and activated neutrophils
In the CF group there were good correlations between the concentration of GSSG and 3-chlorotyrosine (r=0·41, p<0.0004) or GSA (r=0·46, p<0.0001). There were also weaker but significant correlations with MPO, neutrophils, and IL-8. The proportion of GSH present as GSSG showed comparable correlations (tables E1 and E2). Similar data were seen in the non-CF group; however, the weaker correlations in the non-CF group may reflect the lower statistical power due to the smaller number of measurements (table E2).
Protein S-glutathionylation is elevated in CF
To determine whether S-glutathionylation of proteins contributes to the low concentration of GSH in BAL from the children with CF, GSH was liberated from proteins and measured by mass spectrometry. The absolute concentration of GSH attached to proteins in BAL was significantly higher in CF BAL (176 versus 49 nM; p=0.001) than in non-CF BAL (fig. 3a). The percentage of GSH attached to proteins was also significantly higher in CF (11.3 versus 1.1%; p<0.001) (fig 3b). S-glutathionylation in all samples from CF and non-CF disease controls was related to the concentration of MPO (n=37; r=0.42, p=0.001). The absolute concentrations of GSSG (n=37; r=0.72, p<0.001) and GSA (n=37; r=0.82, p<0.001) in these samples were also related to MPO.

{kind=link}
{kind=link}
{kind=link}
S-glutathionylation of proteins in bronchoalveolar lavage (BAL) from children with cystic fibrosis (CF) and without CF (non-CF). a) The absolute concentration of glutathione (GSH) on proteins (protein-SG) and b) its percentage of total GSH in BAL from CF (n=25) and non-CF (n=12) children. The GSH bound to proteins was liberated by reduction and then quantified using liquid chromatography with stable isotope dilution mass spectrometry. Data were analysed using the Mann–Whitney rank sum test on untransformed data.
GSH oxidation is increased during pulmonary infections
To test whether airway infections in CF results in greater oxidation of GSH, the CF group was split into those with and without detectable infections. GSA was significantly elevated in the children with infection (table 2). This coincided with an increase in the percentage of GSH present as GSSG and a corresponding decrease in the percentage of GSH. In line with previous studies, children with pulmonary infections had more neutrophils while NE, MPO, 3-chlorotyrosine, and IL-8 were significantly elevated (table 2).
The contribution of oxidative stress to bronchiectasis in children with CF was also evaluated. Those children with bronchiectasis (n=74) were older (3.7±2.2 versus 1.8±1.7 year; p<0.0001), had higher total cell counts (p=0·027) and higher concentrations of MPO in their BAL (2412±6230 versus 645±1438 ng·mL−1; p=0·043) than those without (n=60). There were no associations between GSH, GSSG, GSA or 3-chlorotyrosine and the presence of bronchiectasis as a binary variable (data not shown).
Discussion
In this study we found that there was a deficit of GSH in the airways of young children with CF compared to disease controls. Our results are in accord with the seminal investigation that highlighted a systemic deficiency of GSH in young adults with CF [11]. Thus, our data demonstrate that GSH is low in the ELF of individuals with CF from an early age. This deficiency will make the lungs of these children more vulnerable to oxidative stress over the course of their disease. Our findings differ from those in an earlier study that showed GSH levels were not significantly lower in infants and young children with CF [25]. The different results probably reflect the greater power in our study and that our method for quantification of GSH is superior to what was available at the time of the previous work.
Since the discovery that patients with CF have a systemic deficiency in GSH, it has been argued strongly that this deficit results directly from an inability of a defective CFTR to transport GSH across the epithelium [8], [26]. However, in this current study it was apparent that the deficit in GSH was partially accounted for by a marked increase in protein S-glutathionylation and was accompanied by hypochlorous acid-dependent oxidation of GSH. Consequently, defects in the CFTR are not solely responsible for the low concentrations of GSH in the ELF. Oxidative stress exerted by activated neutrophils also contributes to the decline in GSH. The combined result of these two effects is that the antioxidant defences in the airways will be impaired. Also, proteins with cysteine residues will be more prone to modification via oxidation and S-glutathionylation.
In previous work we have established that considerable amounts of hypochlorous acid are generated in the airways of children with cystic fibrosis [14], [17]. We reasoned that because hypochlorous acid reacts most rapidly with cysteine residues, it is likely to react with GSH in the ELF. Our finding that GSA was present in BAL and its levels were correlated with the percentage of neutrophils, MPO and 3-chlorotyrosine, confirms our hypothesis that hypochlorous acid oxidises GSH in the airways. This conclusion is reinforced by consistent correlations of GSSG with neutrophils, IL-8, MPO, 3-chlorotyrosine and GSA. However, the precise contribution that hypochlorous acid makes to the oxidation of GSH is difficult to establish because of the dynamic turnover of GSSG. For the majority of children with CF in the present study ∼90% of total free GSH was present as GSH. However, some had a high percentage of GSSG, which suggests that they had experienced considerable oxidative stress and their residual antioxidants defences were severely compromised.
We also showed that proteins in BAL from children with CF are highly S-glutathionylated. Attachment of GSH to proteins was related to MPO and accounted for a substantial proportion of the decreased concentration of unbound GSH in the airways of these children. Mixed disulfides are formed when GSH reacts with oxidised cysteine residues [19]. They can also form via reaction of GSSG with protein thiols but this route is considered to be kinetically incompetent, particularly in the ELF because of the low concentration of GSSG [27]. Thus, the most likely pathway to S-glutathionylation is via oxidation of protein thiols. Hypochlorous acid reacts rapidly with cysteine residues on proteins, and given their high concentration in ELF, they are likely to be major sinks for this oxidant [28]. The protein sulfenic acids formed by hypochlorous acid would couple rapidly with free GSH to produce mixed disulfides. Given that ∼10% of the total GSH in CF BAL was attached to proteins, ∼20% of the decreased concentration of GSH in the CF patients’ airways is most likely to result from oxidation. However, this value will be influenced by the degree of infection and will require further analysis in a larger cohort to confirm.
Despite the relatively small number of CF children with pulmonary infection or bronchiectasis studied, the data from the present study suggests that oxidation of GSH by neutrophil-derived hypochlorous acid is likely to play a part in early CF lung disease. This is because MPO, 3-chlorotyrosine, GSA and the proportion of GSH present as GSSG rose during infection while GSH declined. Previous reports from the AREST CF ESP have demonstrated strong associations between the presence of NE activity in BAL and the presence and progression of structural lung disease, especially bronchiectasis [6], [20]–[22]. In the present study, NE activity was elevated in those with detectable infection (table 2) and tended to be higher in those with bronchiectasis. Thus, both the MPO and NE released during neutrophilic inflammation may be involved in early CF lung disease. They would combine to damage proteins via oxidation and proteolytic digestion. They may also act in concert when hypochlorous acid has depleted GSH to a critical level where it can cause substantial inactivation of α1-antiprotease [29]. At this point NE activity would no longer be blocked by α1-antiprotease and it would be free to digest proteins. The present study was not designed to determine the relative contributions of these neutrophil enzymes to the onset or progression of structural lung disease. However, given that therapeutic strategies can be designed against each pathway, systematic study of the relative contributions of MPO and NE to CF lung disease is warranted.
We propose that when pathogenic bacteria stimulate neutrophils in the CF airways, these inflammatory cells use MPO to generate large amounts of hypochlorous acid. This oxidant will react mainly with cysteine residues on proteins and GSH, depleting GSH and attaching it to proteins or coupling it to form GSSG. Also, based on studies with CFTR knockout mice, the low levels of GSH in CF airways will also make epithelial cells more susceptible to damage by hypochlorous acid [30]. Consequently, targeting the depletion of GSH in CF has therapeutic potential. However, the failure of inhaled GSH to have a clinical impact on CF patients [31] suggests that other strategies should be explored. These include inhibition of MPO, which has the advantage of blocking oxidant production at the source rather than trying to dampen oxidative stress by adding a scavenger, such as GSH, that may not penetrate to the sites of inflammation.
Acknowledgments
AREST CF would like to thank the contributions of Princess Margaret Hospital for Children (Perth, Australia), the Telethon Institute for Child Health Research (Perth, Australia), the Royal Children's Hospital Melbourne (Melbourne, Australia) and the Murdoch Children's Research Institute (Melbourne, Australia). We would also like to thank the participants and their families who contributed to the AREST CF programme.
Footnotes
For editorial comments see page 17.
This article has supplementary material available from www.erj.ersjournals.com
Support statement: Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) receives funding from: the National Health and Medical Research Council (NHMRC) Australia (Centres of Research Excellence #1000896), and the Cystic Fibrosis Foundation Therapeutics, USA (SLY04A0, STICK09A0). The Centre for Free Radical Research in Christchurch receives funding from the Health Research Council of New Zealand.
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
- Received September 30, 2013.
- Accepted February 12, 2014.
- © ERS 2014
References










