





Abstract
The causes of severe childhood asthma are poorly understood. Our aim was to define global patterns of gene expression in children with severe therapy-resistant and controlled asthma.
White blood cells were isolated and the global transcriptome profile was characterised using the Affymetrix Human Gene ST 1.0 chip in children with severe, therapy-resistant asthma (n=17), controlled asthma (n=19) and healthy controls (n=18). Receptor expression was studied in separated leukocyte fractions from asthmatic adults (n=12).
Overall, 1378 genes were differentially expressed between children with severe/controlled asthma and controls. Three significantly enriched Kyoto Encyclopedia of Genes and Genomes pathways were represented: natural killer cell-mediated cytotoxicity (upregulated in controlled asthma); N-glycan biosynthesis (downregulated in severe asthma); and bitter taste transduction receptors (TAS2Rs) (upregulated in severe asthma). Quantitative PCR experiments confirmed upregulation of TAS2Rs in severe asthmatics. TAS2R expression was replicated in leukocytes from adult asthmatics, in which TAS2R agonists also inhibited LPS-induced cytokine release. Significant correlations between expression of TAS2Rs and clinical markers of asthma severity were found in both adults and children.
In conclusion, specific gene expression patterns were observed in children with severe, therapy-resistant asthma. The increased expression of bronchodilatory TAS2Rs suggests a new target for the treatment of asthma.
Introduction
Asthma is a heterogeneous disease that affects millions of adults and children worldwide. Around 5% of children with asthma suffer from chronic symptoms and/or severe exacerbations despite treatment with a variety of drugs, including high doses of inhaled corticosteroids [1]. The term “problematic severe asthma” has been proposed to classify these children, of whom a subgroup suffers from severe, therapy-resistant asthma that probably requires novel therapeutic approaches [2]. Therapy-resistant children should be distinguished from children with other forms of “problematic asthma” that is difficult to treat due to unidentified exacerbating factors, social issues or poor compliance. The causes of severe therapy-resistant childhood asthma are still poorly understood.
Asthma is known to run in families, and studies also suggest that there is a considerable hereditary component for disease severity [3]. To date, there are few genetic studies on therapy-resistant asthma in children, and few studies have explored the gene expression profile that characterises this phenotype [4]. The clinical features of severe asthma, chronic inflammation and subsequent structural changes of the airways, deterioration of lung function and comorbidities [5–7], suggest that genes involved in these processes may be of particular interest. Using a genomics-based approach involving microarray profiling in blood cells collected from children during acute asthma exacerbation and convalescence, gene expression patterns have been characterised and highlight pathways related to, among others, prostaglandin metabolism and innate immunity [8].
Our primary aim was to perform genome-wide expression array analyses on leukocytes obtained from extensively characterised children taking part in a national study on problematic severe asthma [5]. Fresh white blood cells from 17 children with asthma resistant to therapy, 19 patients with controlled persistent asthma and 18 healthy controls were assessed, and our data clearly indicate different gene expression patterns between severe, therapy-resistant asthma and controlled persistent asthma.
One particular novel pathway found to characterise the severe therapy-resistant asthma phenotype was that of bitter taste receptor signalling. This finding was somewhat unexpected as, until recently, bitter taste receptors (TAS2Rs) were thought to be exclusively located on the tongue, where their activation by specific ligands enables our perception of bitter taste. However, Deshpande et al. [9] demonstrated the presence of these receptors in the human lung and, furthermore, that their stimulation induces bronchodilation. A secondary aim of the current study was therefore to perform a more detailed examination of TAS2R expression and function in isolated blood leukocytes. For this purpose, blood was collected from an additional 12 asthmatic subjects and our findings revealed particularly abundant TAS2R expression on lymphocytes, as well as inhibitory effects of TAS2R agonists on pro-inflammatory mediator release. We also found correlations between TAS2R expression and clinical characteristics, suggesting that this pathway may be one important endogenous protective mechanism in severe asthma.
Methods
Study design
Two different cohorts of patients were used in the current study. Genome-wide expression array analyses were performed in mixed blood leukocytes from children taking part in a national study on problematic severe asthma as described [5]. Subsequently, a more detailed examination of bitter taste receptor expression and function was performed on isolated blood leukocytes from an additional 12 adult asthmatic subjects.
Study subjects: children
The nationwide study on problematic severe asthma in Sweden is an observational, multicentre investigation in which school-aged children with problematic severe asthma were compared to age-matched peers with controlled persistent asthma. All patients were required to have been without airway infections or exacerbations of their asthma during a 2-week period prior to examination. The inclusion criteria are presented in table E1, whereas details concerning enrolment are presented elsewhere [5]. Among children who fulfilled the definition of problematic severe asthma, we identified children who were severely resistant to therapy (called severe asthma) based on detailed clinical characterisation (insufficient asthma control despite level 4 treatment, according to the Global Initiative for Asthma (GINA)) [10]. The sample set used in this study comprised 20 children with severe asthma (having an Asthma Control Test® (ACT) score ≤19), 20 children with controlled asthma (selected from the controlled, persistent asthma group, with an ACT score 20–24) and 19 healthy controls recruited at Astrid Lindgren Children’s Hospital, Stockholm, Sweden. The controls had been admitted for elective surgical procedures such as scrotal hernia, and blood samples were collected prior to the planned surgical procedure. Informed consent was obtained from all participating children and their parents and the study was approved by the local ethics committee.
Asthma control test
Self-reported asthma control was assessed using the ACT [11, 12]. A total score of 25–27 points were considered to reflect very well controlled asthma, 20–24 points reflected controlled asthma, and ≤19 points reflected uncontrolled asthma.
Nitric oxide, pulmonary function and assessment of bronchial hyperresponsiveness
Employing a NIOX™ analyser (Aerocrine AB, Solna, Sweden), the fraction of exhaled nitric oxide (FeNO) was measured prior to spirometry, in accordance with international guidelines [13]. Treatment with short- and/or long-acting β2-agonists and/or leukotriene antagonists was withheld for 8, 24 and 72 h, respectively, prior to measurement of baseline pulmonary function and provocation with methacholine. Spirometry was performed with a Vitalograph® 2120 (Vitalograph, Ennis, Ireland), in accordance with published recommendations [14] and using the reference values reported by Polgar and Weng [15]. Bronchial hyperresponsiveness to a challenge with methacholine was assessed using a Spira nebuliser (Spira Respiratory Care Centre, Hämeenlinna, Finland) [16], and results presented as the slope of the dose–response curves (DRSmethacholine) [17].
Blood analyses
Following application of local anaesthesia (EMLA cream; AstraZeneca, Södertälje, Sweden), samples of venous blood were collected. White blood cells and IgE were measured. Atopy was defined as specific IgE >0.35kU·L−1 to inhalant allergens (Phadiatop® (Phadia AB, Uppsala, Sweden), a mixture of cat, dog, horse, birch, timothy, mugwort, Dermatophagoides pteronyssinus and Cladosporium allergens.). However, atopy was not measured in healthy controls as a history of atopy was an exclusion criterion for that group. White blood cells were freshly isolated from the blood, immediately put into RNAlater® (Applied Biosystems/Ambion, Austin, TX, USA) and stored at -20° and -80°C until RNA extraction.
RNA extraction
Total RNA was extracted from white blood cells (buffy coat) using RiboPure™-Blood extraction kit (Applied Biosystems/Ambion) according to the manufacturer's instructions. RNA quality and quantity were assessed using NanoDrop 8000 (Thermo Scientific, Wilmington, DE, USA) and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
Microarray profiling
For transcriptome profiling the Affymetrix Human Gene ST 1.0 chip was used (Affymetrix, Santa Clara, CA, USA). The Gene ST 1.0 chip contains 28 875 genes/transcription sets with a resolution of 26 (median) probes per transcription set. cDNA synthesis and hybridisation were performed according to the manufacturer’s recommendations. A total of 59 hybridisations from 59 subjects were carried out using standard Affymetrix protocols for gene expression (www.affymetrix.com). Experimental data was pre-processed using the statistical software R (www.R-project.org) and Bioconductor (www.bioconductor.org) by implementing the Affy package, and expression values were extracted from the .cel files and normalised using Robust Multi-array Average (justRMA). For quality assessment of the data post-normalisation, the arrayQualityMetrics package was applied [18]. Detected outliers were removed and the data set normalised again. Before further analysis normalisation control genes were filtered away. Data analysis and annotation were performed implementing the limma package and the hugene10sttranscriptcluster library.
In order to validate the results of the microarray profiling, additional quantitative (q) PCR experiments were performed to examine expression of all TAS2Rs found to be differentially expressed between the subject groups. cDNA synthesis and qPCR for 11 commercially available human TAS2Rs were performed exactly as described in the “qPCR for bitter taste receptors” section, below. These experiments were performed in a subset of children: healthy controls (n=18) and children with severe therapy resistant asthma (n=19), for whom sufficient RNA was available. Normally distributed data were compared by unpaired t-test and skewed data using the Mann–Whitney U-test.
Statistics of clinical characteristics in children and microarray data
A two-tailed t-test or ANOVA were used to test for differences in clinical variables between severe asthma, controlled asthma and control groups and the Wilcoxon rank sum test for variables with a skewed distribution (severe asthma versus controlled asthma only). A Chi-squared test was used for differences in categorical variables (sex). A p-value ≤0.05 was regarded as significant. We applied F-statistics to the microarray data as a global test to generate a list of genes differentially expressed in any of the three contrasts: controlled asthma versus healthy controls (controlled asthma–control), severe asthma versus controlled asthma (severe asthma–controlled asthma) and severe asthma versus healthy controls (severe asthma-control). Multiple testing was corrected for using the default Benjamini–Hochberg method and all genes with an adjusted p-value ≤0.05 were regarded as significant, generating a top table of 1378 genes significantly differentially expressed in any of the contrasts. The moderated t-statistics revealed each contrast's contribution to the global test of differential expression.
Clustering and functional enrichment analysis
Unsupervised hierarchic clustering and heatmap were generated implementing the gplots package using all 1378 significant genes implicated by the F-statistics. Functional annotation and enrichment analysis were performed to determine whether more genes than expected by chance were from a given biological pathway or process. Biological process (BP_FAT) and pathways (Kyoto Encyclopaedia of Genes and Genomes) were assessed using DAVID 6.7 (http://david.abcc.ncifcrf.gov) [19, 20]. Enrichment was considered significant if the Benjamini–Hochberg adjusted p-value was ≤0.05 relative to the Affymetrix Human Gene ST 1.0 background. All microarray data are available in the Gene Expression Omnibus repository (www.ncbi.nlm.nih.gov/projects/geo/) (accession no. GSE27011).
Study subjects: adults
In order to investigate the expression and function of bitter taste receptors on isolated blood leukocyte fractions, blood was collected from an additional 12 adult patients with intermittent allergic asthma, documented airway hyperresponsiveness to methacholine and a positive skin prick test to pollen or animal dander. Subject characteristics are shown in table E2. These subjects were recruited from the outpatient clinic at the Dept of Respiratory Medicine and Allergy, Karolinska University Hospital (Stockholm, Sweden), and all subjects provided written consent to participate in the study, which was approved by the local ethics committee.
Isolation of blood leukocyte fractions
Peripheral venous blood was collected into Vacutainer® tubes (Beckton Dickinson, Franklin Lakes, NJ, USA) containing heparin. Following dextran sedimentation of erythrocytes, Lymphoprep® (Axis-Shield, Dundee, UK) density gradient separation was performed to separate a granulocyte-rich pellet and peripheral blood mononuclear-rich interphase. Neutrophils and monocytes were isolated by positive selection using CD16 and CD14 MACS® microbeads (Miltenyi Biotech, BergischGladbach, Germany), respectively, and lymphocytes by negative selection following the removal of monocytes according to the manufacturer’s instructions. These preparations yielded a purity of >95%.
Quantitative PCR for bitter taste receptors
RNA was extracted using the RNeasy® Mini kit (Qiagen, Hilden, Germany) with on-column deoxyribonuclease digestion according to the manufacturer’s instructions. Briefly, fresh cells were homogenised in the RLT lysis buffer supplied, and stored at -80°C before proceeding with the RNA extraction protocol. RNA concentration and quality was assessed using a NanoDrop 8000. cDNA synthesis was carried out using the QuantiTect Reverse Transcription kit (Qiagen, Hamburg, Germany) according to the manufacturer’s instructions. qPCR was carried out using an ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) with TaqMan® reagents (Life Technologies/Invitrogen, Carlsbad, CA, USA). 11 commercially available TaqMan® primers for human TAS2Rs (hTAS2R4, 5, 10, 13, 14, 19, 20, 31, 45, 46 and 50) and the housekeeping gene glyceraldehyde-3-phosphatase dehydrogenase (GAPDH) were used. As negative controls, PCR reactions were performed using water or products of the cDNA reaction performed in the absence of the reverse transcriptase enzyme. Gene expression relative to that of GAPDH was calculated according to the 2−ΔCT formula.
Effect of TAS2R agonists on leukocyte function
In order to examine the effect of TAS2R activation on leukocyte function, the TAS2R agonists chloroquine and denatonium were applied to a standardised assay known to increase pro-inflammatory mediator release by blood leukocytes. Briefly, whole blood was incubated with 100 μg·mL−1 lipopolysaccharide (LPS) for 24 h, with or without 10, 100 or 1000 μM of the bitter taste receptor agonist or relevant vehicle control. Plasma was then removed and stored at -80°C prior to measurement of pro-inflammatory cytokines and prostaglandin (PG)E2. A panel of 17 cytokines (interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-17A, granulocyte colony stimulating factor (G-CSF), granulocyte/macrophage colony stimulating factor (GM-CSF), interferon (IFN)-γ, monocyte chemotactic protein (MCP)-1, macrophage inflammatory protein-1β and tumour necrosis factor (TNF)-α) were assayed in serum (diluted 1:4), using a Bio-Plex Pro™ 17-plex kit with a Luminex® analyser (Bio-Rad Laboratories, Hercules, CA, USA). PGE2 levels were measured by enzyme immunoassay (Cayman Chemical, Ann Arbor, MI, USA).
Statistics
The majority of datasets showing TAS2R expression in isolated blood leukocytes were normally distributed according to the Kolmogorov–Smirnov test and were therefore compared using a paired t-test. Correlations between receptor expression and clinical characteristics were examined using Pearson's test. A p-value <0.05 was accepted as statistically significant.
Results
Identification of genes subdividing severe asthma, controlled asthma and controls
The transcriptomes of 59 subjects were assayed on Affymetrix Human Gene ST 1.0 expression arrays. After pre-processing and quality control of the post-hybridised arrays, five samples were regarded as outliers and removed. The remaining sample set of 17 severe asthma, 19 controlled asthma and 18 healthy controls were used for downstream analyses. Application of a linear modelling approach and F-test statistics identified a top table of 1378 transcription clusters/genes that were significantly differentially expressed in one, or several, of the controlled asthma–control, severe asthma–controlled asthma or severe asthma–control contrasts (see table E3). T-test statistics were applied to identify which of the contrasts contributed to the overall significance, and Figure 1 illustrates this distribution. Based on these 1378 genes, a large number seemed to uniquely differentiate the severe asthma group from the controlled asthma (n=351 genes) and control (n=315 genes) groups, whereas fewer genes differentiated the controlled asthma from the control group (n=149). 355 genes were also shared between the severe asthma–control and severe asthma–controlled asthma contrasts, which supported the view that severe asthma was characterised by a unique set of genes compared with the other two groups.

A Venn diagram illustrating the distribution of differentially expressed genes between children with severe asthma (SA), controlled asthma (CA) and controls. The numbers in the circles depict the number of genes uniquely differentially expressed or shared between the contrasts: CA versus healthy controls (CA–control), SA versus CA (SA–CA) and SA versus healthy controls (SA–control). The number of downregulated or upregulated genes in each of the contrasts is indicated below the diagram.
The clinical data of the three groups (severe asthma, controlled asthma and controls) are presented in table 1. A significant increase was seen for severe asthma compared to controlled asthma for bronchial hyperresponsiveness (DRSmethacholine, p=0.01), and controls were slightly younger than children with severe asthma and controlled asthma. However, there were no significant differences seen for forced expiratory volume in 1 s (FEV1), total white blood cell count, eosinophils, neutrophils, FeNO, atopy or total IgE. No statistics were performed on ACT or inhaled corticosteroids, of which a derivation in score or dose, respectively, was an inclusion criterion for the expression analysis.
An unsupervised hierarchical clustering and heatmap of the normalised raw intensity of the 1378 differentially expressed genes illustrated a more refined grouping of the pre-defined severe asthma, controlled asthma and controls (fig. 2). The column dendrogram separated the majority of severe asthma from controlled asthma and control individuals (cluster I and II, respectively), and also separated the majority of control individuals from severe asthma and controlled asthma (all but two controls found in IIb). Controlled asthma patients were found both in IIa and IIb.
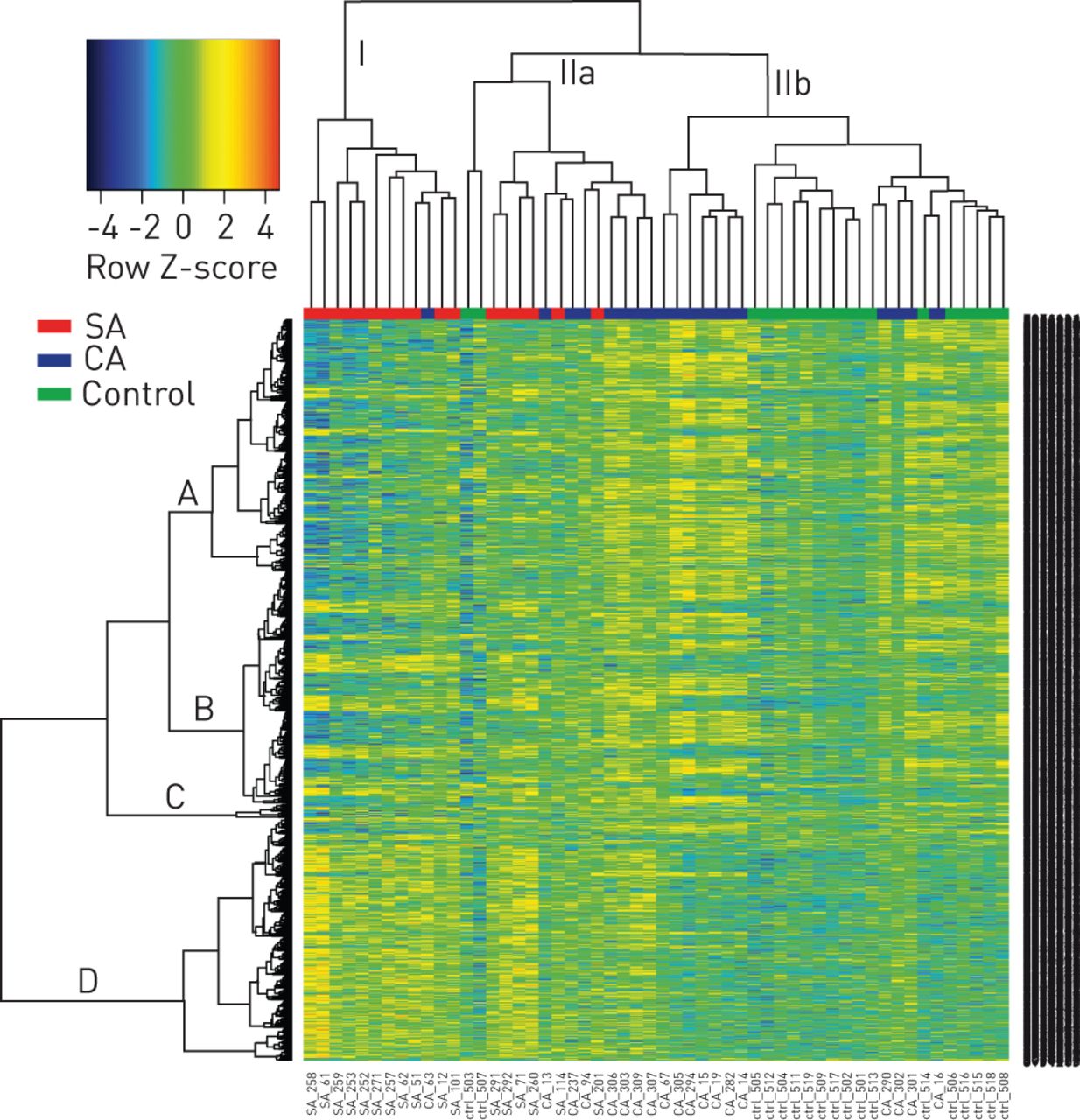
Global gene expression patterns in children with severe asthma (SA), controlled asthma (CA) and healthy controls. Unsupervised hierarchical clustering and heatmap illustrates each individual's expression pattern in all 1378 significant (p≤0.05), differentially expressed genes. The column dendrogram separates the subjects into three main clusters (I, IIa and IIb). The row dendrogram represents gene clusters crudely divided into clusters A, B, C and D. Normalised gene expression is indicated by the row Z-score where yellow–red represents upregulated genes and blue represents downregulated genes.
Novel pathways and processes in asthma
Pathways enrichment analyses using the DAVID database were performed utilising two separate approaches. First, all 1378 differentially expressed genes in the top table were used, revealing three significantly enriched Kyoto Encyclopedia of Genes and Genomes pathways: taste transduction (mostly upregulated in severe asthma); natural killer (NK) cell-mediated cytotoxicity (upregulated in controlled asthma); and N-glycan biosynthesis (downregulated in severe asthma) (table 2). Secondly, we made use of the dendrogram row clusters comprising the heatmap (fig. 2). We crudely dissected out four blocks from the row dendrogram (A, B, C and D in Figure 2) and performed functional annotation on these clusters (table 2). Cluster A contained the NK cell-mediated cytotoxicity pathway, but also revealed two additional pathways: T-cell receptor signalling and citrate cycle; all of these being upregulated in controlled asthma. Cluster D contained the bitter taste receptor pathway, being upregulated in all severe asthma and some controlled asthma patients with a remarkably improved p-value (from 0.03 using the top table genes to 4.2×10−10).
To look for additional functional enriched groupings we performed gene ontology enrichment analysis. In severe asthma versus healthy controls, sensory perception of taste was significantly enriched. Other significant biological processes related to metabolic processes and immune processes were also identified. For a complete list see table E3. Out of the total list of 1378 differentially expressed genes, 15% of the transcript clusters were annotated as known, or, in most cases, novel noncoding RNA (ncRNA), mostly represented by small nuclear RNA, small nucleolar RNA, ribosomal RNA, miscellaneous RNA and microRNA (see table E4.). The majority of ncRNAs were found to be upregulated in severe asthma compared to controlled asthma or controls.
Confirmation of TAS2R expression in children by qPCR
Expression of mRNA for 11 different TAS2R (4, 5, 10, 13, 14, 19, 20, 31, 45, 46 and 50) was confirmed by qPCR in mixed blood leukocyte samples from healthy children and children with severe therapy-resistant asthma (fig. E1). In general, the expression of most bitter taste receptors was higher in children with severe asthma compared to the healthy controls, and these differences reached statistical significance for TAS2R13 (p=0.028), TAS2R14 (p=0.009) and TAS2R19 (p=0.037).
Co-expression of genetically associated asthma genes
Against our list of 1378 differentially expressed genes we compared a list of 97 previously recognised asthma candidate genes, identified either through well-replicated association studies or through genome-wide association studies [21]. 12 genes were present in both lists (table 3). Most genes were upregulated in controlled asthma alone with three exceptions: RORA, which was upregulated in both severe and controlled asthma, and PEX19 and MYB, which were significantly downregulated in severe asthma alone. Functional annotation revealed no shared pathways, but several of the genes (RORA, TBX21, ADRB2, IL2RB, PDE4D, MYB and PAFAH2) were involved in the same biological processes, such as positive regulation of various metabolic processes, regulation of cell death and leukocyte activation.
Relative expression of TAS2Rs in isolated blood leukocytes
Expression of mRNA for the 11 commercially available human bitter taste receptors relative to GAPDH in mixed leukocytes, lymphocytes, neutrophils and monocytes is shown in Figure 3. Expression of all bitter taste receptors was significantly greatest in the lymphocyte population compared to monocytes. Expression of the TAS2R was also greater in lymphocytes than neutrophils, but for TAS2R50, TAS2R45, TAS2R31 and TAS2R5 these differences did not reach statistical significance. In lymphocytes, the relative expression of TAS2R10 was higher than that of the other TAS2Rs examined. The expression of TAS2R5 on mixed blood leukocytes showed a significant inverse correlation with lung function in these subjects, both FEV1 % predicted (p=0.011, Pearson's correlation coefficient= -0.83) and forced vital capacity % pred (p=0.029). Interestingly, in the asthmatic children, TAS2R5 mRNA expression in mixed blood leukocytes (log2 TAS2R5 expression derived from the Affymetrix Human Gene ST 1.0 chip) also showed a statistically significant correlation with bronchial hyperresponsiveness to methacholine, measured as the slope of the dose–response curve (p=0.008, Pearson's correlation coefficient=0.47).

Expression of human bitter taste receptors (TAS2R) in isolated leukocytes from adult asthmatics. The relative expression of TAS2R (4, 5, 10, 13, 14, 19, 20, 31, 45, 46 and 50) compared to glyceraldehyde-3-phosphatase dehydrogenase (GAPDH), as measured by quantitative PCR, was examined in mixed leukocytes and isolated lymphocytes, monocytes and neutrophils (n=12). Results are shown as mean±sem and expression levels of the different TAS2Rs in the different cell populations were compared by t-test. CT: cycle threshold. *: p<0.05.
Effect of TAS2R agonists on LPS-induced inflammatory mediator release
In order to examine the possible role of TAS2R activation on blood leukocyte function, the effect of the prototype TAS2R agonists chloroquine and denatonium (10–1000 μM) on LPS-induced inflammatory mediator release was examined. Results showed that both chloroquine and denatonium significantly inhibited the release of TNF-α, IL-13 and MCP-1. Chloroquine also inhibited the release of IL-1β, IL-2, IL-4, IL-5, IL-10, IL-17, G-CSF, GM-CSF, IFN-γ and PGE2 in a dose-dependent manner (fig. 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of bitter taste receptor (TAS2R) agonists on lipopolysaccharide (LPS)-induced pro-inflammatory cytokine and prostaglandin (PG)E2 release. Blood samples from 12 asthmatic adults were incubated for 24 h, either unstimulated (C) or stimulated with LPS (100 μg·mL−1), in the absence (LPS) or presence of chloroquine or denatonium (10–1000 μM). Serum levels of 17 cytokines (interleukin (IL)-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-17, tumour necrosis factor (TNF)-α, granulocyte colony stimulating factor (G-CSF), granulocyte/macrophage colony stimulating factor (GM-CSF), interferon (IFN)-γ, monocyte chemotactic protein (MCP)-1 and macrophage inflammatory protein (MIP)-1α) were analysed by luminex and PGE2 by enzyme immunoassay. Results for IL-6, IL-8 and MIP-1α are not shown as these were present at levels above the detection limit of the assay used. *: p<0.05 compared to LPS alone.
Discussion
Using genome-wide expression arrays to analyse the transcriptomes of 54 asthmatic patients and healthy controls, we identified a separation in gene expression between severe therapy-resistant asthma and well-controlled persistent asthma. Whereas traditional immune-related functions, such as T-cell receptor signalling and NK cell-mediated cytotoxicity, were found to be upregulated in controlled asthmatics, we could not observe such a pattern in the severe asthmatics. Instead, we saw a profound enrichment and upregulation of TAS2Rs, which highlighted a pathway previously not associated with severe therapy-resistant asthma. These findings were further strengthened by our qPCR analyses, also showing increased TAS2R expression in these subjects.
In this study we looked at the genome-wide expression pattern in whole white blood cells with the aim of finding unique expression profiles that could separate the predefined patient groups. By using a global approach that identified genes differentially expressed in any of the contrasts, we were able to identify 1378 significantly (p≤0.05) altered genes. Unsupervised hierarchical clustering and heatmap of the gene expression in all samples (fig. 2) separated most of the severe asthmatics (cluster I) from controlled asthmatics and the healthy control group (cluster II). Although the expression patterns could classify the patient groups rather well, it is evident that other biological and physiological parameters also contributed to these complex asthma phenotypes.
Thus, we observed a profound enrichment and upregulation of the TAS2R pathway in leukocytes from the severe asthmatics, which became even more significant from the cluster analyses in the heatmap dendrogram. Until recently, TAS2Rs were not known to have any role in asthma. However, TAS2Rs have now been connected to the respiratory system by four independent studies. Shah et al. [22] showed that TAS2R were expressed on the motile cilia of human airway epithelial cells and that bitter compounds increased the intracellular Ca2+ concentration and stimulated ciliary beat frequency. The motile cells are responsible for propelling mucus and harmful substances out of the lung and it has been shown that viral infections and cigarette smoke causes a loss of these cilia and might disrupt the defensive mechanism [23]. Very recent data suggest that a specific TAS2R (38) is involved in the innate immune defence of upper airways and that variation in this gene contributes to individual differences in susceptibility to respiratory infection [24]. Deshpande et al. [9] found that the TAS2Rs were expressed in human airway smooth muscle (ASM) and that bitter tastants caused relaxation of isolated ASM and dilation of airways. Inhaled bitter tastants also caused decreased airway obstruction in a mouse model of asthma. We have recently confirmed and extended these observations by documenting that bitter taste receptor agonists also induce relaxation of guinea pig trachea, and that they may also relax maximally contracted bronchi that are unresponsive to β-agonists [25]. In addition, our present findings of inhibitory effects of two TAS2R agonists on leukocyte function suggest that activation of bitter taste receptors may have anti-inflammatory actions as well. Taken together, these studies suggest a protective role for the TAS2Rs in asthma. Our study is the first to report upregulation of TAS2Rs in human white blood cells in patients suffering from severe therapy-resistant asthma.
In order to examine in more detail which white blood cells express bitter taste receptors we performed qPCR for 11 TAS2R in isolated lymphocyte, monocyte and neutrophil fractions from a subset of 12 well-characterised adult asthmatic subjects. Results showed that expression of all bitter taste receptors was greatest in the lymphocyte fraction, and furthermore that hTAS2R10 appeared to be the most abundantly expressed. Although hTAS2R10 was not differentially expressed between the different groups of children it was included in the panel of receptors examined by qPCR, as TAS2R10 was found to be one of the most highly expressed bitter taste receptors on airway smooth muscle in the study by Deshpande et al [9]. While these findings relate to a different group of patients, specific differences in TAS2R expression on lymphocytes may underlie the differences observed between the controlled and severe asthmatics, as TAS2R expression in lymphocytes was often similar to that observed in mixed leukocyte samples. As similar white blood cell counts were observed between the children with controlled and severe asthma (table 1), the observed TAS2R upregulation is unlikely to be due to differences in leukocyte numbers. Exactly which lymphocyte subsets, and the biological relevance of TAS2R expression on these cells, should be the focus of future investigations. Our initial experiments have shown that the TAS2R agonists chloroquine and denatonium are able to inhibit the release of several pro-inflammatory cytokines and eicosanoid release from blood leukocytes (fig. 4), suggesting that these agents may have anti-inflammatory as well as bronchodilatory properties; however, more detailed functional studies are still required. The broader profile of activity of chloroquine compared with denatonium, however, confirms recent findings in airway smooth muscle [25].
The pathway analyses also demonstrated that T-cell related pathways, such as NK T-cell mediated cytotoxicity, were upregulated in controlled asthmatics, whereas N-glycan biosynthesis was downregulated in the severe asthma group. The role of NK cells in asthma has been debated and studies have both shown a vast upregulation of NK T-cells in bronchoalveolar lavage fluid from asthmatic patients [26], as well as no increase at all [27]. Murine models have supported the role of NK T-cells in asthma by showing that NK T-cell deficient mice do not develop, or show impaired, allergen-induced airway hyperreactivity [28, 29]. Our results point towards a role for NK T-cells in asthma. The N-glycan biosynthesis pathway is responsible for the production of glycans that couple to proteins and lipids on the cell membrane. Alterations in the distribution of glycoproteins at the cell surface have been suggested to contribute to many chronic human diseases, including autoimmunity [30].
A great number of the genes identified, primarily in severe asthma, were annotated as ncRNA, or novel ncRNA. Recent genome-wide expression studies have shown that the human genome produces many thousands of these regulatory, non-protein-coding RNAs and that they play roles in virtually all developmental processes, but primarily in transcriptional and post-transcriptional regulation [31]. Subsequently, ncRNAs have been linked to various human diseases such as cancer, central nervous system disorders and cardiovascular disease [31]. The role of ncRNA in asthma, and in particular therapy-resistant asthma, is, however, unclear and remains a target for further investigation.
12 out of 97 well-replicated asthma genes were found among the 1378 differentially expressed genes (table 3). Thus, these genes have been associated with asthma at the DNA as well as the RNA level, which supports their role in the pathogenesis of disease. Most of the genes were upregulated only in controlled asthma (PEX19, RORA and MYB being exceptions). This might be due to the fact that most association studies, as well as genome-wide association studies, consulted to generate the asthma gene list were conducted on combined patient materials consisting mostly of mild/moderate asthmatics.
Our study has limitations. First, we measured gene expression patterns in peripheral blood cells (mainly leukocytes), which are only one of several possible target cells in controlled/severe asthma. However, the immune system is a large component in the aetiology of asthma. Some other studies have measured gene expression levels from cells obtained from bronchial alveolar lavage or bronchial biopsies [32–34]. A direct comparison with these studies is therefore difficult to make; however, combining data performed in various relevant tissues might help us paint a more complete picture of asthma. Our pathway analyses were stringent and only explored known pathways, which is always a limiting factor. A liberal inclusion of differentially expressed genes (p≤0.05, regardless of fold change) was compensated with a stricter approach for the functional annotation. This was done in an attempt to increase power but reduce false positives. A larger study size would have been preferred, but almost 60 study subjects was nevertheless a rather large number for genome-wide expression analyses. Although history of atopy was an exclusion criterion for the healthy control group, we cannot completely rule out that some control children had specific IgE >0.35 kU·L−1 (since this was not measured).
In conclusion, the current study provides new insight into the characteristics of severe therapy-resistant asthma in children. It is of note that this sufficiently powered genome-wide transcriptome analysis was possible only because of the accessibility to a group of asthmatic patients who were exceptionally well phenotyped. Our data in the controlled asthmatics supported differential expression of known pathways, such as T-cell related networks. However, the novel finding was that the study revealed specific upregulation of pathways previously not associated with asthma in the severe asthma group, for example N-glycan biosynthesis and, in particular, bitter taste transduction. The increased expression of bronchodilatory bitter taste receptors and the initial indications of anti-inflammatory responses mediated by these receptors certainly warrant further explorations of their potential as a new target for asthma therapy. Finally, it may be more than coincidental that beneficial therapeutic effects have been reported for treatment with chloroquine in asthma [35].
Acknowledgments
We would like to thank all participating children in the Swedish National Study on Severe Asthma and their parents, all adult asthma patients at the Clinical Asthma and Allergy Research Laboratory (Karolinska University Hospital Huddinge, Stockholm, Sweden), the Bioinformatics and Expression Analysis Affymetrix core facility at NOVUM, Karolinska Institute, Stockholm, and Ingegerd Fransson (Karolinska Institute, Stockholm, Sweden) for technical assistance, as well as the staff at the Clinical Asthma and Allergy Research Laboratory and The Global Allergy and Asthma European Network (GA2LEN).
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support statement: This work was supported by the Centre for Allergy Research, Karolinska Institutet, Konsul Th. C. Bergh’s Foundation, the Freemason Child House Foundation in Stockholm, the Swedish Asthma and Allergy Association’s Research Foundation, the Paediatric Research Foundation of Astrid Lindgren Children’s Hospital, the Swedish Heart–Lung Foundation, Novo Nordisk Foundation, Swedish Foundation for Strategic Research, the Stockholm County Council Research Funds (ALF), Vinnova (CIDaT), the KI-AZ-TSC-JRP project, and research funds of the University of Helsinki and Swedish Medical Research Council (VR-M). E. Melén is supported by a grant from Riksbankens Jubileumsfond, Erik Rönnberg's scholarship for research on early childhood diseases. A. James and M. Kupczyk are post-doctoral fellows in the Bernard Osher Initiative for Research on Severe Asthma at Karolinska Institutet.
Conflict of interest: None declared.
- Received May 15, 2012.
- Accepted November 13, 2012.
- ©ERS 2013
References










